
In genetics, dominance is the phenomenon of one variant (allele) of a gene on a chromosome masking or overriding the effect of a different variant of the same gene on the other copy of the chromosome. The first variant is termed dominant and the second is called recessive. This state of having two different variants of the same gene on each chromosome is originally caused by a mutation in one of the genes, either new or inherited. The terms autosomal dominant or autosomal recessive are used to describe gene variants on non-sex chromosomes (autosomes) and their associated traits, while those on sex chromosomes (allosomes) are termed X-linked dominant, X-linked recessive or Y-linked; these have an inheritance and presentation pattern that depends on the sex of both the parent and the child. Since there is only one copy of the Y chromosome, Y-linked traits cannot be dominant or recessive. Additionally, there are other forms of dominance, such as incomplete dominance, in which a gene variant has a partial effect compared to when it is present on both chromosomes and co-dominance, in which different variants on each chromosome both show their associated traits.

Hemoglobinopathy is the medical term for a group of inherited blood disorders and diseases that primarily affect red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

The globins are a superfamily of heme-containing globular proteins, involved in binding and/or transporting oxygen. These proteins all incorporate the globin fold, a series of eight alpha helical segments. Two prominent members include myoglobin and hemoglobin. Both of these proteins reversibly bind oxygen via a heme prosthetic group. They are widely distributed in many organisms.

Haptoglobin is the protein that in humans is encoded by the HP gene. In blood plasma, haptoglobin binds with high affinity to free hemoglobin released from erythrocytes, and thereby inhibits its deleterious oxidative activity. Compared to Hp, hemopexin binds to free heme. The haptoglobin-hemoglobin complex will then be removed by the reticuloendothelial system.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time.
DNA banking is the secure, long term storage of an individual’s genetic material. DNA is most commonly extracted from blood, but can also be obtained from saliva and other tissues. DNA banks allow for conservation of genetic material and comparative analysis of an individual's genetic information. Analyzing an individual's DNA can allow scientists to predict genetic disorders, as used in preventive genetics or gene therapy, and prove that person's identity, as used in the criminal justice system. There are multiple methods for testing and analyzing genetic information including restriction fragment length polymorphism (RFLP) and polymerase chain reactions (PCR).
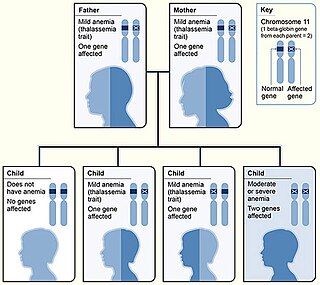
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Marco Antonio Zago is a Brazilian physician and prominent medical scientist, who is active in the fields of hereditary diseases of the blood, molecular basis of cancer and human population genetics. Aside from working directly as scientes, he has been president of the Brazilian National Research Council (CNPq) and dean of the University of São Paulo. He's now the president of the São Paulo Research Foundation (Fapesp).
A locus control region (LCR) is a long-range cis-regulatory element that enhances expression of linked genes at distal chromatin sites. It functions in a copy number-dependent manner and is tissue-specific, as seen in the selective expression of β-globin genes in erythroid cells. Expression levels of genes can be modified by the LCR and gene-proximal elements, such as promoters, enhancers, and silencers. The LCR functions by recruiting chromatin-modifying, coactivator, and transcription complexes. Its sequence is conserved in many vertebrates, and conservation of specific sites may suggest importance in function. It has been compared to a super-enhancer as both perform long-range cis regulation via recruitment of the transcription complex.
The human β-globin locus is composed of five genes located on a short region of chromosome 11, responsible for the creation of the beta parts of the oxygen transport protein Haemoglobin. This locus contains not only the beta globin gene but also delta, gamma-A, gamma-G, and epsilon globin. Expression of all of these genes is controlled by single locus control region (LCR), and the genes are differentially expressed throughout development.
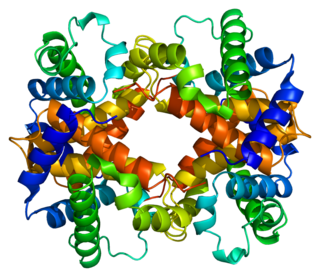
Hemoglobin subunit beta is a globin protein, coded for by the HBB gene, which along with alpha globin (HBA), makes up the most common form of haemoglobin in adult humans, hemoglobin A (HbA). It is 147 amino acids long and has a molecular weight of 15,867 Da. Normal adult human HbA is a heterotetramer consisting of two alpha chains and two beta chains.

Hemoglobin variants are different types of hemoglobin molecules, by different combinations of its subunits and/or mutations thereof. Hemoglobin variants are a part of the normal embryonic and fetal development. They may also be pathologic mutant forms of hemoglobin in a population, caused by variations in genetics. Some well-known hemoglobin variants, such as sickle-cell anemia, are responsible for diseases and are considered hemoglobinopathies. Other variants cause no detectable pathology, and are thus considered non-pathological variants.

Krueppel-like factor 1 is a protein that in humans is encoded by the KLF1 gene. The gene for KLF1 is on the human chromosome 19 and on mouse chromosome 8. Krueppel-like factor 1 is a transcription factor that is necessary for the proper maturation of erythroid cells.

Hemoglobin subunit delta is a protein that in humans is encoded by the HBD gene.

Probable G-protein coupled receptor 124 is a protein that in humans is encoded by the GPR124 gene. It is a member of the adhesion-GPCR family of receptors. Family members are characterized by an extended extracellular region with a variable number of protein domains coupled to a TM7 domain via a domain known as the GPCR-Autoproteolysis INducing (GAIN) domain.
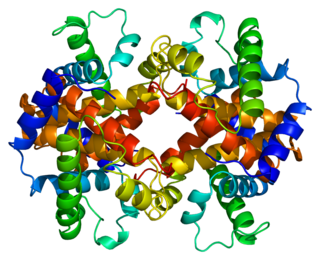
Hemoglobin subunit epsilon is a protein that in humans is encoded by the HBE1 gene.
Human genetic resistance to malaria refers to inherited changes in the DNA of humans which increase resistance to malaria and result in increased survival of individuals with those genetic changes. The existence of these genotypes is likely due to evolutionary pressure exerted by parasites of the genus Plasmodium which cause malaria. Since malaria infects red blood cells, these genetic changes are most common alterations to molecules essential for red blood cell function, such as hemoglobin or other cellular proteins or enzymes of red blood cells. These alterations generally protect red blood cells from invasion by Plasmodium parasites or replication of parasites within the red blood cell.

Hemoglobin Lepore syndrome is typically an asymptomatic hemoglobinopathy, which is caused by an autosomal recessive genetic mutation. The Hb Lepore variant, consisting of two normal alpha globin chains (HBA) and two delta-beta globin fusion chains which occurs due to a "crossover" between the delta (HBD) and beta globin (HBB) gene loci during meiosis and was first identified in the Lepore family, an Italian-American family, in 1958. There are three varieties of Hb Lepore, Washington, Baltimore and Hollandia. All three varieties show similar electrophoretic and chromatographic properties and hematological findings bear close resemblance to those of the beta-thalassemia trait; a blood disorder that reduces the production of the iron-containing protein hemoglobin which carries oxygen to cells and which may cause anemia.

HBS1 like translational GTPase is a protein that in humans is encoded by the HBS1L gene.