This article has multiple issues. Please help improve it or discuss these issues on the talk page . (Learn how and when to remove these template messages)
|
This article has multiple issues. Please help improve it or discuss these issues on the talk page . (Learn how and when to remove these template messages)
|
The knowledge of protein structures may facilitate and improve the annotation of protein function and the characterization of protein binding partners and binding sites. A database and server IBIS (Inferred Biomolecular Interaction Server, www.ncbi.nlm.nih.gov/Structure/ibis/ibis.cgi) [1] [2] developed at NCBI, National Institutes of Health, reports, predicts and integrates multiple types of conserved interactions for proteins. It provides tools to analyze biomolecular interactions observed in a given protein structure together with the complex set of interactions inferred from its close homologs. IBIS identifies and predicts proteins' interaction partners together with the locations of the corresponding binding sites on the protein query. It provides annotations of binding sites for protein-protein, protein- small molecule, protein - nucleic acid, protein - peptide and protein - ion interactions.
IBIS also allows the mapping of a biomolecular interaction network for any given organism, human interactome derived from structural complexes is available at ftp://ftp.ncbi.nih.gov/pub/mmdb/humanIntNw/.

To focus on biologically relevant binding sites, IBIS clusters similar binding sites found in homologous proteins based on the sites’ conservation of sequence and structure. Binding sites which appear evolutionarily conserved among non-redundant sets of homologous proteins are given higher priority. After binding sites are clustered, Position Specific Score Matrices (PSSMs) are constructed from the corresponding binding site alignments. Together with other measures, the PSSMs are subsequently used to rank binding sites to assess how well they match the query, and to gauge the biological relevance of binding sites with respect to the query.
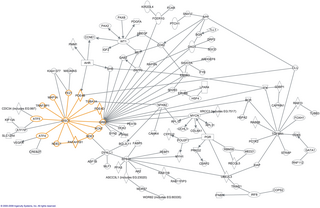
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules but can also describe sets of indirect interactions among genes.

UniProt is a freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. It is maintained by the UniProt consortium, which consists of several European bioinformatics organisations and a foundation from Washington, DC, United States.
InterPro is a database of protein families, domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
Protein–protein interaction prediction is a field combining bioinformatics and structural biology in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding protein–protein interactions is important for the investigation of intracellular signaling pathways, modelling of protein complex structures and for gaining insights into various biochemical processes.
Structural and physical properties of DNA provide important constraints on the binding sites formed on surfaces of DNA-binding proteins. Characteristics of such binding sites may be used for predicting DNA-binding sites from the structural and even sequence properties of unbound proteins. This approach has been successfully implemented for predicting the protein–protein interface. Here, this approach is adopted for predicting DNA-binding sites in DNA-binding proteins. First attempt to use sequence and evolutionary features to predict DNA-binding sites in proteins was made by Ahmad et al. (2004) and Ahmad and Sarai (2005). Some methods use structural information to predict DNA-binding sites and therefore require a three-dimensional structure of the protein, while others use only sequence information and do not require protein structure in order to make a prediction.
The Biomolecular Object Network Databank is a bioinformatics databank containing information on small molecule structures and interactions. The databank integrates a number of existing databases to provide a comprehensive overview of the information currently available for a given molecule.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.
The Conserved Domain Database (CDD) is a database of well-annotated multiple sequence alignment models and derived database search models, for ancient domains and full-length proteins.
The Molecular Modeling Database (MMDB) is a database of experimentally determined three-dimensional biomolecular structures and hosted by the National Center for Biotechnology Information.

UPF0172 protein FAM158A, also known as c14orf122 or CGI112, is a protein that in humans is encoded by the FAM158A gene located on chromosome 14q11.2.
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.
Megf8 also known as Multiple Epidermal Growth Factor-like Domains 8, is a protein coding gene that encodes a single pass membrane protein, known to participate in developmental regulation and cellular communication. It is located on chromosome 19 at the 49th open reading frame in humans (19q13.2). There are two isoform constructs known for MEGF8, which differ by a 67 amino acid indel. The isoform 2 splice version is 2785 amino acids long, and predicted to be 296.6 kdal in mass. Isoform 1 is composed of 2845 amino acids and predicted to weigh 303.1 kdal. Using BLAST searches, orthologs were found primarily in mammals, but MEGF8 is also conserved in invertebrates and fishes, and rarely in birds, reptiles, and amphibians. A notably important paralog to multiple epidermal growth factor-like domains 8 is ATRNL1, which is also a single pass transmembrane protein, with several of the same key features and motifs as MEGF8, as indicated by Simple Modular Architecture Research Tool (SMART) which is hosted by the European Molecular Biology Laboratory located in Heidelberg, Germany. MEGF8 has been predicted to be a key player in several developmental processes, such as left-right patterning and limb formation. Currently, researchers have found MEGF8 SNP mutations to be the cause of Carpenter syndrome subtype 2.

DEP Domain Containing Protein 1B also known as XTP1, XTP8, HBV XAg-Transactivated Protein 8, [formerly referred to as BRCC3] is a human protein encoded by a gene of similar name located on chromosome 5.

Intermediate filament family orphan 1 is a protein that in humans is encoded by the IFFO1 gene. IFFO1 has uncharacterized function and a weight of 61.98 kDa. IFFO1 proteins play an important role in the cytoskeleton and the nuclear envelope of most eukaryotic cell types.
C6orf222 is a protein that in humans is encoded by the C6orf222 gene (6p21.31). C6orf222 is conserved in mammals, birds and reptiles with the most distant ortholog being the green sea turtle, Chelonia mydas. The C6orf222 protein contains one mammalian conserved domain: DUF3293. The protein is also predicted to contain a BH3 domain, which has predicted conservation in distant orthologs from the clade Aves.
ProBiS is a computer software which allows prediction of binding sites and their corresponding ligands for a given protein structure. Initially ProBiS was developed as a ProBiS algorithm by Janez Konc and Dušanka Janežič in 2010 and is now available as ProBiS server, ProBiS CHARMMing server, ProBiS algorithm and ProBiS plugin. The name ProBiS originates from the purpose of the software itself, that is to predict for a given Protein structure Binding Sites and their corresponding ligands.
Transmembrane Protein 217 is a protein encoded by the gene TMEM217. TMEM217 has been found to have expression correlated with the lymphatic system and endothelial tissues and has been predicted to have a function linked to the cytoskeleton.

SHLD1 or shieldin complex subunit 1 is a gene on chromosome 20. The C20orf196 gene encodes an mRNA that is 1,763 base pairs long, and a protein that is 205 amino acids long.

Chromosome 1 open reading frame 112, is a protein that in humans is encoded by the C1orf112 gene, and is located at position 1q24.2. C1orf112 encodes for seventeen variants of mRNA, fifteen of which are functional proteins. C1orf112 has a determined precursor molecular weight of 96.6 kDa and an isoelectric point of 5.62. C1orf112 has been experimentally determined to localize to the mitochondria, although it does not contain a mitochondrial targeting sequence.