
The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group donates a hydrogen bond to the backbone C=O group of the amino acid located three or four residues earlier along the protein sequence.

The β-sheet is a common motif of regular secondary structure in proteins. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in formation of the protein aggregates and fibrils observed in many human diseases, notably the amyloidoses such as Alzheimer's disease.

Proline (symbol Pro or P) is a proteinogenic amino acid that is used in the biosynthesis of proteins. It contains an α-amino group (which is in the protonated NH2+ form under biological conditions), an α-carboxylic acid group (which is in the deprotonated −COO− form under biological conditions), and a side chain pyrrolidine, classifying it as a nonpolar (at physiological pH), aliphatic amino acid. It is non-essential in humans, meaning the body can synthesize it from the non-essential amino acid L-glutamate. It is encoded by all the codons starting CC (CCU, CCC, CCA, and CCG).
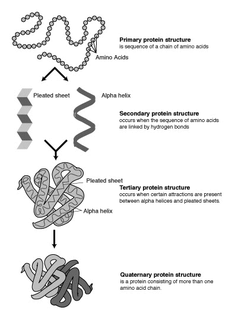
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its folding and its secondary and tertiary structure from its primary structure. Structure prediction is fundamentally different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by bioinformatics and theoretical chemistry; it is highly important in medicine and biotechnology. Every two years, the performance of current methods is assessed in the CASP experiment. A continuous evaluation of protein structure prediction web servers is performed by the community project CAMEO3D.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.

A pi helix is a type of secondary structure found in proteins. Although once thought to be rare, short π-helices are found in 15% of known protein structures and are believed to be an evolutionary adaptation derived by the insertion of a single amino acid into an α-helix. Because such insertions are highly destabilizing, the formation of π-helices would tend to be selected against unless it provided some functional advantage to the protein. π-helices therefore are typically found near functional sites of proteins.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.
Conformational entropy is the entropy associated with the number of conformations of a molecule. The concept is most commonly applied to biological macromolecules such as proteins and RNA, but also be used for polysaccharides and other molecules. To calculate the conformational entropy, the possible conformations of the molecule may first be discretized into a finite number of states, usually characterized by unique combinations of certain structural parameters, each of which has been assigned an energy. In proteins, backbone dihedral angles and side chain rotamers are commonly used as parameters, and in RNA the base pairing pattern may be used. These characteristics are used to define the degrees of freedom. The conformational entropy associated with a particular structure or state, such as an alpha-helix, a folded or an unfolded protein structure, is then dependent on the probability of the occupancy of that structure.
Loop modeling is a problem in protein structure prediction requiring the prediction of the conformations of loop regions in proteins with or without the use of a structural template. Computer programs that solve these problems have been used to research a broad range of scientific topics from ADP to breast cancer. Because protein function is determined by its shape and the physiochemical properties of its exposed surface, it is important to create an accurate model for protein/ligand interaction studies. The problem arises often in homology modeling, where the tertiary structure of an amino acid sequence is predicted based on a sequence alignment to a template, or a second sequence whose structure is known. Because loops have highly variable sequences even within a given structural motif or protein fold, they often correspond to unaligned regions in sequence alignments; they also tend to be located at the solvent-exposed surface of globular proteins and thus are more conformationally flexible. Consequently, they often cannot be modeled using standard homology modeling techniques. More constrained versions of loop modeling are also used in the data fitting stages of solving a protein structure by X-ray crystallography, because loops can correspond to regions of low electron density and are therefore difficult to resolve.
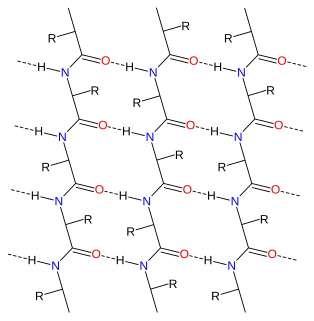
Alpha sheet is an atypical secondary structure in proteins, first proposed by Linus Pauling and Robert Corey in 1951. The hydrogen bonding pattern in an alpha sheet is similar to that of a beta sheet, but the orientation of the carbonyl and amino groups in the peptide bond units is distinctive; in a single strand, all the carbonyl groups are oriented in the same direction on one side of the pleat, and all the amino groups are oriented in the same direction on the opposite side of the sheet. Thus the alpha sheet accumulates an inherent separation of electrostatic charge, with one edge of the sheet exposing negatively charged carbonyl groups and the opposite edge exposing positively charged amino groups. Unlike the alpha helix and beta sheet, the alpha sheet configuration does not require all component amino acid residues to lie within a single region of dihedral angles; instead, the alpha sheet contains residues of alternating dihedrals in the traditional right-handed (αR) and left-handed (αL) helical regions of Ramachandran space. Although the alpha sheet is only rarely observed in natural protein structures, it has been speculated to play a role in amyloid disease and it was found to be a stable form for amyloidogenic proteins in molecular dynamics simulations. Alpha sheets have also been observed in X-ray crystallography structures of designed peptides.
Peptide plane flipping is a type of conformational change that can occur in proteins by which the dihedral angles of adjacent amino acids undergo large-scale rotations with little displacement of the side chains. The plane flip is defined as a rotation of the dihedral angles φ,ψ at amino acids i and i+1 such that the resulting angles remain in structurally stable regions of Ramachandran space. The key requirement is that the sum of the ψi angle of residue i and the φi+1 angle of residue i+1 remain roughly constant; in effect, the flip is a crankshaft move about the axis defined by the Cα-C¹ and N-Cα bond vectors of the peptide group, which are roughly parallel. As an example, the type I and type II beta turns differ by a simple flip of the central peptide group of the turn.
Hydrophobicity scales are values that define relative hydrophobicity of amino acid residues. The more positive the value, the more hydrophobic are the amino acids located in that region of the protein. These scales are commonly used to predict the transmembrane alpha-helices of membrane proteins. When consecutively measuring amino acids of a protein, changes in value indicate attraction of specific protein regions towards the hydrophobic region inside lipid bilayer.
Protein backbone fragment libraries have been used successfully in a variety of structural biology applications, including homology modeling, de novo structure prediction, and structure determination. By reducing the complexity of the search space, these fragment libraries enable more rapid search of conformational space, leading to more efficient and accurate models.

CheShift-2 is an application created to compute 13Cα and 13Cβ protein chemical shifts and to validate protein structures. It is based on quantum mechanics computations of 13Cα and 13Cβchemical shift as a function of the torsional angles of the 20 amino acids.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of >15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality and identifying residue “outliers”. The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR’s output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.
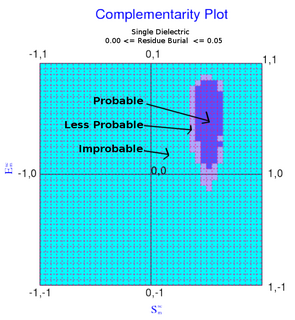
The complementarity plot (CP) is a graphical tool for structural validation of atomic models for both folded globular proteins and protein-protein interfaces. It is based on a probabilistic representation of preferred amino acid side-chain orientation, analogous to the preferred backbone orientation of Ramachandran plots). It can potentially serve to elucidate protein folding as well as binding. The upgraded versions of the software suite is available and maintained in github for both folded globular proteins as well as inter-protein complexes. The software is included in the bioinformatic tool suites OmicTools and Delphi tools.



