
Gardner's syndrome is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon. The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.
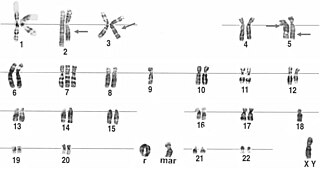
A ring chromosome is an aberrant chromosome whose ends have fused together to form a ring. Ring chromosomes were first discovered by Lilian Vaughan Morgan in 1926. A ring chromosome is denoted by the symbol r in human genetics and R in Drosophila genetics. Ring chromosomes may form in cells following genetic damage by mutagens like radiation, but they may also arise spontaneously during development.
Acromicric dysplasia is an extremely rare inherited disorder characterized by abnormally short hands and feet, growth retardation and delayed bone maturation leading to short stature. Most cases have occurred randomly for no apparent reason (sporadically). However, autosomal dominant inheritance has not been ruled out.

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.
Catel–Manzke syndrome is a rare genetic disorder characterized by distinctive abnormalities of the index fingers; the classic features of Pierre Robin syndrome; occasionally with additional physical findings.

Gray platelet syndrome (GPS), or platelet alpha-granule deficiency, is a rare congenital autosomal recessive bleeding disorder caused by a reduction or absence of alpha-granules in blood platelets, and the release of proteins normally contained in these granules into the marrow, causing myelofibrosis. The name derives from the initial observation of gray appearance of platelets with a paucity of granules on blood films from a patient with a lifelong bleeding disorder.
Schinzel–Giedion syndrome (SGS) is a congenital neurodegenerative terminal syndrome. It was first described in 1978 by Albert Schinzel (1944–) and Andreas Giedion (1925–) as a syndrome with severe midface retraction, skull anomalies, renal anomalies (hydronephrosis) and other anomalies. Babies born with Schinzel–Giedion syndrome have severe mental retardation, growth retardation and global developmental delay.
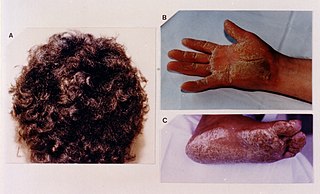
Naxos disease is a cutaneous condition characterized by a palmoplantar keratoderma. The prevalence of the syndrome is up to 1 in every 1000 people in the Greek islands.
Morgagni-Stewart-Morel syndrome is a condition with a wide range of associated endocrine problems including: diabetes mellitus, diabetes insipidus, and hyperparathyroidism. Other signs and symptoms include headaches, vertigo, hirsutism, menstrual disorder, galactorrhoea, obesity, depression, and seizures. Thickening of the inner table of the frontal part of the skull a usually benign condition known as hyperostosis frontalis interna. The syndrome was first described in 1765. It is named after the Italian anatomist and pathologist Giovanni Battista Morgagni, the British neurologist Roy Mackenzie Stewart, and the Swiss psychiatrist Ferdinand Morel.
Majeed syndrome is an inherited skin disorder characterized by chronic recurrent multifocal osteomyelitis, congenital dyserythropoietic anemia and a neutrophilic dermatosis. It is classified as an autoinflammatory bone disorder. The condition is found in people with two defective copies of the LPIN2 gene. LPIN2 encodes lipin-2 which is involved in lipid metabolism. The pathogenesis of this mutation with the clinical manifestations has not been elucidated.
Lyngstadaas syndrome, also known as severe dental aberrations in familial steroid dehydrogenase deficiency, is a rare autosomal recessive liver disease involving an enzyme deficiency and dental anomalies. The disease is named after the Norwegian professor Ståle Petter Lyngstadaas.

Gordon syndrome, or distal arthrogryposis type 3, is a rare genetic disorder characterized by cleft palate and congenital contractures of the hands and feet.
Hereditary gingival fibromatosis (HGF), also known as idiopathic gingival hyperplasia, is a rare condition of gingival overgrowth. HGF is characterized as a benign, slowly progressive, nonhemorrhagic, fibrous enlargement of keratinized gingiva. It can cover teeth in various degrees, and can lead to aesthetic disfigurement. Fibrous enlargement is most common in areas of maxillary and mandibular tissues of both arches in the mouth. Phenotype and genotype frequency of HGF is 1:175,000 where males and females are equally affected but the cause is not entirely known. It mainly exists as an isolated abnormality but can also be associated with a multi-system syndrome.
Heart-hand syndromes are a group of rare diseases that manifest with both heart and limb deformities.
Mietens syndrome is a autosomal recessive disorder first described by Mietens and Weber. The condition is named after a German physician named Carl Mietens.
Nathalie syndrome is a rare genetic developmental defect during embryogenesis disorder and is thought to be hereditary.
Wells-Jankovic syndrome is a rare neurologic disorder characterized by spastic paraparesis presents in late childhood along with hearing loss.
van Den Bosch syndrome is a rare X-linked syndrome like intellectual disability. It may be caused by a small X-chromosome deletion.

Pai syndrome, also known as Median cleft of the upper lip-corpus callosum lipoma-midline facial cutaneous polyps syndrome, is a very rare genetic disorder which is characterized by nervous system, cutaneous, ocular, nasal and bucal anomalies with facial dysmorphisms.
Spondylometaphyseal dysplasia with cone-rod dystrophy is a rare genetic disorder characterized by spondylometaphyseal dysplasia, neonatal growth delays, and cone-rod dystrophy-associated progressive vision loss. Only 18 patients from families in the United States, the United Kingdom, Japan, and Brazil have been described to date. This condition is caused by autosomal recessive mutations in the PCYT1A gene, located in chromosome 3.