Adducted thumb syndrome recessive form is a rare disease affecting multiple systems causing malformations of the palate, thumbs, and upper limbs. The name Christian syndrome derives from Joe. C. Christian, the first person to describe the condition. Inheritance is believed to be autosomal recessive, caused by mutation in the CHST14 gene.

Popliteal pterygium syndrome (PPS) is an inherited condition affecting the face, limbs, and genitalia. The syndrome goes by a number of names including the popliteal web syndrome and, more inclusively, the facio-genito-popliteal syndrome. The term PPS was coined by Gorlin et al. in 1968 on the basis of the most unusual anomaly, the popliteal pterygium.

Duane-radial ray syndrome, also known as Okihiro Syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.

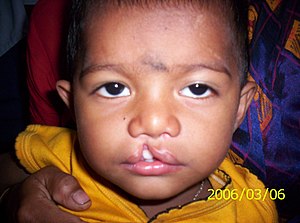
Ectrodactyly–ectodermal dysplasia–cleft syndrome, or EEC, and also referred to as EEC syndrome and split hand–split foot–ectodermal dysplasia–cleft syndrome is a rare form of ectodermal dysplasia, an autosomal dominant disorder inherited as a genetic trait. EEC is characterized by the triad of ectrodactyly, ectodermal dysplasia, and facial clefts. Other features noted in association with EEC include vesicoureteral reflux, recurrent urinary tract infections, obstruction of the nasolacrimal duct, decreased pigmentation of the hair and skin, missing or abnormal teeth, enamel hypoplasia, absent punctae in the lower eyelids, photophobia, occasional cognitive impairment and kidney anomalies, and conductive hearing loss.
RAPADILINO syndrome is an autosomal recessive disorder characterized by:

Raine syndrome (RNS), also called osteosclerotic bone dysplasia, is a rare autosomal recessive congenital disorder characterized by craniofacial anomalies including microcephaly, noticeably low set ears, osteosclerosis, a cleft palate, gum hyperplasia, a hypoplastic nose, and eye proptosis. It is considered to be a lethal disease, and usually leads to death within a few hours of birth. However, a recent report describes two studies in which children with Raine syndrome have lived to 8 and 11 years old, so it is currently proposed that there is a milder expression that the phenotype can take.
Hay–Wells syndrome is one of at least 150 known types of ectodermal dysplasia. These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.
Rosselli–Gulienetti syndrome, also known as Zlotogora–Ogur syndrome and Bowen–Armstrong syndrome, is a type of congenital ectodermal dysplasia syndrome. The syndrome is relatively rare and has only been described in a few cases.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.
Acrofrontofacionasal dysostosis is an extremely rare disorder, characterized by intellectual disability, short stature, hypertelorism, broad notched nasal tip, cleft lip/palate, postaxial camptobrachypolysyndactyly, fibular hypoplasia, and anomalies of foot structure.

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.
Goldberg–Shprintzen is a very rare connective tissue condition associated with mutations in KIAA1279 gene which encodes KIF-binding protein (KBP), a protein that may interact with microtubules and actin filaments. KBP may play a key role in cytoskeleton formation and neurite growth.

Ectrodactyly, split hand, or cleft hand involves the deficiency or absence of one or more central digits of the hand or foot and is also known as split hand/split foot malformation (SHFM). The hands and feet of people with ectrodactyly (ectrodactyls) are often described as "claw-like" and may include only the thumb and one finger with similar abnormalities of the feet.
Floating–Harbor syndrome, also known as Pelletier–Leisti syndrome, is a rare disease with fewer than 50 cases described in the literature. It is usually diagnosed in early childhood and is characterized by the triad of proportionate short stature with delayed bone age, characteristic facial appearance, and delayed speech development. Although its cause is unknown, it is thought to result from genetic mutation, and diagnosis is established by the presence of a heterozygous SRCAP mutation in those with clinical findings of FHS.
Neu–Laxova syndrome is a rare autosomal recessive disorder characterized by severe intrauterine growth restriction and multiple congenital malformations. Neu–Laxova syndrome is a very severe disorder, leading to stillbirth or death shortly after birth. It was first described by Dr. Richard Neu in 1971 and Dr. Renata Laxova in 1972 as a lethal disorder in siblings with multiple malformations. Neu–Laxova syndrome is an extremely rare disorder with less than 100 cases reported in medical literature.
Opitz G/BBB syndrome, also known as Opitz syndrome, G syndrome or BBB syndrome, is a rare genetic disorder that will affect physical structures along the midline of the body. The letters G and BBB represent the last names of the families that were first diagnosed with the disorder, while Opitz is the last name of the doctor that first described the signs and symptoms of the disease. There are two different forms of Optiz G/BBB syndrome: x-linked (recessive) syndrome and dominant autosomal syndrome. However, both result in common physical deformities, although their pattern of inheritance may differ. Several other names for the disease(s) are no longer used. These include hypospadias-dysphagia syndrome, Opitz-Frias syndrome, telecanthus with associated abnormalities, and hypertelorism-hypospadias syndrome.
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.
Mandibulofacial dysostosis with microcephaly syndrome, also known as growth delay-intellectual disability-mandibulofacial dysostosis-microcephaly-cleft palate syndrome, mandibulofacial dysostosis, guion-almeida type, or simply as MFDM syndrome is a rare genetic disorder which is characterized by developmental delays, intellectual disabilities, and craniofacial dysmorphisms.
Holoprosencephaly-ectrodactyly-cleft lip/palate syndrome, also simply known as Hartsfield syndrome, is a rare genetic disorder characterized by the presence of variable holoprosencephaly, ectrodactyly, cleft lip and palate, alongside generalized ectodermal abnormalities. Additional findings include endocrine anomalies and developmental delays.