Immunodeficiency, also known as immunocompromisation, is a state in which the immune system's ability to fight infectious diseases and cancer is compromised or entirely absent. Most cases are acquired ("secondary") due to extrinsic factors that affect the patient's immune system. Examples of these extrinsic factors include HIV infection and environmental factors, such as nutrition. Immunocompromisation may also be due to genetic diseases/flaws such as SCID.
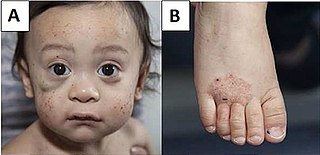
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954. The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.

Béguez-Chédiak–Higashi syndrome (CHS) is a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis. The decrease in phagocytosis results in recurrent pyogenic infections, albinism, and peripheral neuropathy.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections.
CTLA-4 or CTLA4, also known as CD152, is a protein receptor that functions as an immune checkpoint and downregulates immune responses. CTLA-4 is constitutively expressed in regulatory T cells but only upregulated in conventional T cells after activation – a phenomenon which is particularly notable in cancers. It acts as an "off" switch when bound to CD80 or CD86 on the surface of antigen-presenting cells.
Hypogammaglobulinemia is a problem with the immune system in which not enough gamma globulins are produced in the blood. This results in a lower antibody count, which impairs the immune system, increasing risk of infection. Hypogammaglobulinemia may result from a variety of primary genetic immune system defects, such as common variable immunodeficiency, or it may be caused by secondary effects such as medication, blood cancer, or poor nutrition, or loss of gamma globulins in urine, as in nonselective glomerular proteinuria. Patients with hypogammaglobulinemia have reduced immune function; important considerations include avoiding use of live vaccines, and take precautionary measures when traveling to regions with endemic disease or poor sanitation such as receiving immunizations, taking antibiotics abroad, drinking only safe or boiled water, arranging appropriate medical cover in advance of travel, and ensuring continuation of any immunoglobulin infusions needed.
Common variable immunodeficiency (CVID) is an immune disorder characterized by recurrent infections and low antibody levels, specifically in immunoglobulin (Ig) types IgG, IgM and IgA. Generally symptoms include high susceptibility to foreign invaders, chronic lung disease, and inflammation and infection of the gastrointestinal tract. It affects males and females equally. The condition can be found in children or teens but is generally not diagnosed or recognized until adulthood. The average age of diagnosis is between 20-50. However, symptoms vary greatly between people. "Variable" refers to the heterogeneous clinical manifestations of this disorder, which include recurrent bacterial infections, increased risk for autoimmune disease and lymphoma, as well as gastrointestinal disease. CVID is a lifelong disease.
Immune reconstitution inflammatory syndrome (IRIS) is a condition seen in some cases of AIDS or immunosuppression, in which the immune system begins to recover, but then responds to a previously acquired opportunistic infection with an overwhelming inflammatory response that paradoxically makes the symptoms of infection worse.
Bare lymphocyte syndrome type II is a rare recessive genetic condition in which a group of genes called major histocompatibility complex class II are not expressed. The result is that the immune system is severely compromised and cannot effectively fight infection.
Cat skin disorders are among the most common health problems in cats. Skin disorders in cats have many causes, and many of the common skin disorders that afflict people have a counterpart in cats. The condition of a cat's skin and coat can also be an important indicator of its general health. Skin disorders of cats vary from acute, self-limiting problems to chronic or long-lasting problems requiring life-time treatment. Cat skin disorders may be grouped into categories according to the causes.
An immune disorder is a dysfunction of the immune system. These disorders can be characterized in several different ways:
Lipopolysaccharide-responsive and beige-like anchor protein is a protein that in humans is encoded by the LRBA gene.
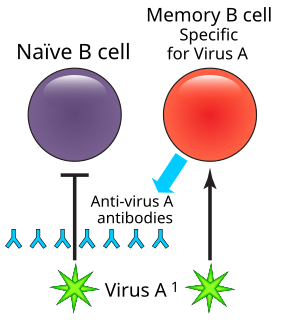
Humoral immune deficiencies are conditions which cause impairment of humoral immunity, which can lead to immunodeficiency. It can be mediated by insufficient number or function of B cells, the plasma cells they differentiate into, or the antibody secreted by the plasma cells. The most common such immunodeficiency is inherited selective IgA deficiency, occurring between 1 in 100 and 1 in 1000 persons, depending on population. They are associated with increased vulnerability to infection, but can be difficult to detect in the absence of infection.
DOCK8 deficiency, also called DOCK8 immunodeficiency syndrome, is the autosomal recessive form of hyperimmunoglobulin E syndrome, a genetic disorder characterized by elevated immunoglobulin E levels, eosinophilia, and recurrent infections with staphylococcus and viruses. It is caused by a mutation in the DOCK8 gene.
LRBA deficiency is a rare genetic disorder of the immune system. This disorder is caused by a mutation in the gene LRBA. LRBA stands for “lipopolysaccharide (LPS)-responsive and beige-like anchor protein”. This condition is characterized by autoimmunity, lymphoproliferation, and immune deficiency. It was first described by Gabriela Lopez-Herrera from University College London in 2012. Investigators in the laboratory of Dr. Michael Lenardo at National Institute of Allergy and Infectious Diseases, the National Institutes of Health and Dr. Michael Jordan at Cincinnati Children’s Hospital Medical Center later described this condition and therapy in 2015.
Granulomatous–lymphocytic interstitial lung disease (GLILD) is a lung complication of common variable immunodeficiency disorders (CVID). It is seen in approximately 15% of patients with CVID. It has been defined histologically as the presence of (non-caseating) granuloma and lymphoproliferation in the lung. However, as GLILD is often associated with other auto-immune features such as splenomegaly, adenopathy and cytopenias, a definition based on abnormalities on lung imaging together with evidence of granulomatous inflammation elsewhere has also been employed.
Nuclear factor-kappa B Essential Modulator (NEMO) deficiency syndrome is a rare type of primary immunodeficiency disease that has a highly variable set of symptoms and prognoses. It mainly affects the skin and immune system but has the potential to affect all parts of the body, including the lungs, urinary tract and gastrointestinal tract. It is a monogenetic disease caused by mutation in the IKBKG gene. NEMO is the modulator protein in the IKK inhibitor complex that, when activated, phosphorylates the inhibitor of the NF-κB transcription factors allowing for the translocation of transcription factors into the nucleus.
Suranjith Seneviratne is a doctor from Sri Lanka who practices in allergy and immunology.
CHAI disease is a rare genetic disorder of the immune system that illustrates the role of CTLA-4 in cell signaling. CHAI stands for “Autoimmune lymphoproliferative syndrome due to CTLA4 haplo-insufficiency.” The disease is characterized by variable combination of enteropathy, hypogammaglobulinemia, recurrent respiratory infections, granulomatous lymphocytic interstitial lung disease, lymphocytic infiltration of non-lymphoid organs, autoimmune thrombocytopenia or neutropenia, autoimmune hemolytic anemia and lymphadenopathy. It is closely linked to LATIAE disease. Investigators in the laboratory of Dr. Michael Lenardo, National Institute of Allergy and Infectious Diseases at the National Institutes of Health first described this condition in 2018.
