Related Research Articles

In genetics, complementary DNA (cDNA) is DNA synthesized from a single-stranded RNA template in a reaction catalyzed by the enzyme reverse transcriptase. cDNA is often used to express a specific protein in a cell that does not normally express that protein, or to sequence or quantify mRNA molecules using DNA based methods. cDNA that codes for a specific protein can be transferred to a recipient cell for expression, often bacterial or yeast expression systems. cDNA is also generated to analyze transcriptomic profiles in bulk tissue, single cells, or single nuclei in assays such as microarrays, qPCR, and RNA-seq.

The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. A synthetic primer may also be referred to as an oligo, short for oligonucleotide. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesizes the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.

A reverse transcriptase (RT) is an enzyme used to generate complementary DNA (cDNA) from an RNA template, a process termed reverse transcription. Reverse transcriptases are used by viruses such as HIV and hepatitis B to replicate their genomes, by retrotransposon mobile genetic elements to proliferate within the host genome, and by eukaryotic cells to extend the telomeres at the ends of their linear chromosomes. Contrary to a widely held belief, the process does not violate the flows of genetic information as described by the classical central dogma, as transfers of information from RNA to DNA are explicitly held possible.
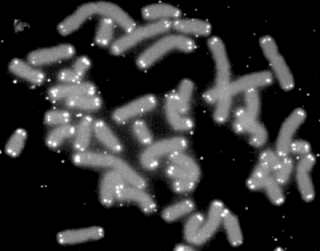
A telomere is a region of repetitive nucleotide sequences associated with specialized proteins at the ends of linear chromosomes. Telomeres are a widespread genetic feature most commonly found in eukaryotes. In most, if not all species possessing them, they protect the terminal regions of chromosomal DNA from progressive degradation and ensure the integrity of linear chromosomes by preventing DNA repair systems from mistaking the very ends of the DNA strand for a double-strand break.

Reverse transcription polymerase chain reaction (RT-PCR) is a laboratory technique combining reverse transcription of RNA into DNA and amplification of specific DNA targets using polymerase chain reaction (PCR). It is primarily used to measure the amount of a specific RNA. This is achieved by monitoring the amplification reaction using fluorescence, a technique called real-time PCR or quantitative PCR (qPCR). Confusion can arise because some authors use the acronym RT-PCR to denote real-time PCR. In this article, RT-PCR will denote Reverse Transcription PCR. Combined RT-PCR and qPCR are routinely used for analysis of gene expression and quantification of viral RNA in research and clinical settings.

Thermus aquaticus is a species of bacteria that can tolerate high temperatures, one of several thermophilic bacteria that belong to the Deinococcota phylum. It is the source of the heat-resistant enzyme Taq DNA polymerase, one of the most important enzymes in molecular biology because of its use in the polymerase chain reaction (PCR) DNA amplification technique.
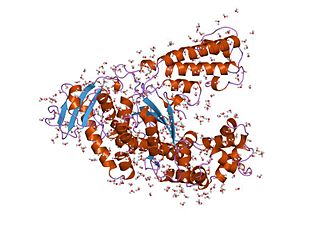
Taq polymerase is a thermostable DNA polymerase I named after the thermophilic eubacterial microorganism Thermus aquaticus, from which it was originally isolated by Chien et al. in 1976. Its name is often abbreviated to Taq or Taq pol. It is frequently used in the polymerase chain reaction (PCR), a method for greatly amplifying the quantity of short segments of DNA.

Dideoxynucleotides are chain-elongating inhibitors of DNA polymerase, used in the Sanger method for DNA sequencing. They are also known as 2',3' because both the 2' and 3' positions on the ribose lack hydroxyl groups, and are abbreviated as ddNTPs.
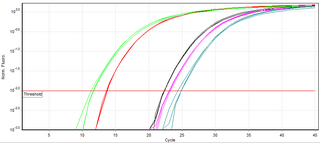
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.
Pfu DNA polymerase is an enzyme found in the hyperthermophilic archaeon Pyrococcus furiosus, where it functions to copy the organism's DNA during cell division. In the laboratory setting, Pfu is used to amplify DNA in the polymerase chain reaction (PCR), where the enzyme serves the central function of copying a new strand of DNA during each extension step.
TaqMan probes are hydrolysis probes that are designed to increase the specificity of quantitative PCR. The method was first reported in 1991 by researcher Kary Mullis at Cetus Corporation, and the technology was subsequently developed by Hoffmann-La Roche for diagnostic assays and by Applied Biosystems for research applications.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
The polymerase chain reaction (PCR) is a commonly used molecular biology tool for amplifying DNA, and various techniques for PCR optimization which have been developed by molecular biologists to improve PCR performance and minimize failure.
Digital polymerase chain reaction is a biotechnological refinement of conventional polymerase chain reaction methods that can be used to directly quantify and clonally amplify nucleic acids strands including DNA, cDNA, or RNA. The key difference between dPCR and traditional PCR lies in the method of measuring nucleic acids amounts, with the former being a more precise method than PCR, though also more prone to error in the hands of inexperienced users. A "digital" measurement quantitatively and discretely measures a certain variable, whereas an “analog” measurement extrapolates certain measurements based on measured patterns. PCR carries out one reaction per single sample. dPCR also carries out a single reaction within a sample, however the sample is separated into a large number of partitions and the reaction is carried out in each partition individually. This separation allows a more reliable collection and sensitive measurement of nucleic acid amounts. The method has been demonstrated as useful for studying variations in gene sequences — such as copy number variants and point mutations — and it is routinely used for clonal amplification of samples for next-generation sequencing.
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
A primer dimer (PD) is a potential by-product in the polymerase chain reaction (PCR), a common biotechnological method. As its name implies, a PD consists of two primer molecules that have attached (hybridized) to each other because of strings of complementary bases in the primers. As a result, the DNA polymerase amplifies the PD, leading to competition for PCR reagents, thus potentially inhibiting amplification of the DNA sequence targeted for PCR amplification. In quantitative PCR, PDs may interfere with accurate quantification.
Hot start PCR is a modified form of conventional polymerase chain reaction (PCR) that reduces the presence of undesired products and primer dimers due to non-specific DNA amplification at room temperatures. Many variations and modifications of the PCR procedure have been developed in order to achieve higher yields; hot start PCR is one of them. Hot start PCR follows the same principles as the conventional PCR - in that it uses DNA polymerase to synthesise DNA from a single stranded template. However, it utilizes additional heating and separation methods, such as inactivating or inhibiting the binding of Taq polymerase and late addition of Taq polymerase, to increase product yield as well as provide a higher specificity and sensitivity. Non-specific binding and priming or formation of primer dimers are minimized by completing the reaction mix after denaturation. Some ways to complete reaction mixes at high temperatures involve modifications that block DNA polymerase activity in low temperatures, use of modified deoxyribonucleotide triphosphates (dNTPs), and the physical addition of one of the essential reagents after denaturation.
Gibson assembly is a molecular cloning method that allows for the joining of multiple DNA fragments in a single, isothermal reaction. It is named after its creator, Daniel G. Gibson, who is the chief technology officer and co-founder of the synthetic biology company, Telesis Bio. The technology is more efficient than manual plasmid genetic recombination methods, but remains expensive as it is still under patent.
Recombinase polymerase amplification (RPA) is a single tube, isothermal alternative to the polymerase chain reaction (PCR). By adding a reverse transcriptase enzyme to an RPA reaction it can detect RNA as well as DNA, without the need for a separate step to produce cDNA,. Because it is isothermal, RPA can use much simpler equipment than PCR, which requires a thermal cycler. Operating best at temperatures of 37–42 °C and still working, albeit more slowly, at room temperature means RPA reactions can in theory be run quickly simply by holding a tube. This makes RPA an excellent candidate for developing low-cost, rapid, point-of-care molecular tests. An international quality assessment of molecular detection of Rift Valley fever virus performed as well as the best RT-PCR tests, detecting less concentrated samples missed by some PCR tests and an RT-LAMP test. RPA was developed and launched by TwistDx Ltd., a biotechnology company based in Cambridge, UK.
References
- ↑ "PCR Master Mix". Sigma-Aldrich. Merck.
- ↑ "GoTaq® G2 Master Mixes | PCR Master Mix". www.promega.com.
- ↑ "PCR Master Mix | Bio-Rad". www.bio-rad.com. Bio Rad.
- ↑ "PCR Master Mixes | Chai". www.chaibio.com. Chai.
- ↑ Yang, Jianxin; Kemps-Mols, Berit; Spruyt-Gerritse, Marijke; Anholts, Jacqueline; Claas, Frans; Eikmans, Michael (4 June 2016). "The source of SYBR green master mix determines outcome of nucleic acid amplification reactions". BMC Research Notes. 9 (1). doi: 10.1186/s13104-016-2093-4 . PMC 4893258 . PMID 27259280.
- ↑ Jiménez, Karen M.; Forero, Diego A. (5 April 2018). "Effect of master mixes on the measurement of telomere length by qPCR". Molecular Biology Reports. 45 (4): 633–638. doi:10.1007/s11033-018-4175-y. S2CID 254833450.
- ↑ Lin, Jue; Smith, Dana L.; Esteves, Kyle; Drury, Stacy (January 2019). "Telomere length measurement by qPCR – Summary of critical factors and recommendations for assay design". Psychoneuroendocrinology. 99: 271–278. doi:10.1016/j.psyneuen.2018.10.005. PMC 6363640 . PMID 30343983.
| | This molecular biology article is a stub. You can help Wikipedia by expanding it. |