Related Research Articles

An epigenome consists of a record of the chemical changes to the DNA and histone proteins of an organism; these changes can be passed down to an organism's offspring via transgenerational stranded epigenetic inheritance. Changes to the epigenome can result in changes to the structure of chromatin and changes to the function of the genome.

RNA editing is a molecular process through which some cells can make discrete changes to specific nucleotide sequences within an RNA molecule after it has been generated by RNA polymerase. It occurs in all living organisms and is one of the most evolutionarily conserved properties of RNAs. RNA editing may include the insertion, deletion, and base substitution of nucleotides within the RNA molecule. RNA editing is relatively rare, with common forms of RNA processing not usually considered as editing. It can affect the activity, localization as well as stability of RNAs, and has been linked with human diseases.
Cross-linking and immunoprecipitation is a method used in molecular biology that combines UV crosslinking with immunoprecipitation in order to identify RNA binding sites of proteins on a transcriptome-wide scale, thereby increasing our understanding of post-transcriptional regulatory networks. CLIP can be used either with antibodies against endogenous proteins, or with common peptide tags or affinity purification, which enables the possibility of profiling model organisms or RBPs otherwise lacking suitable antibodies.
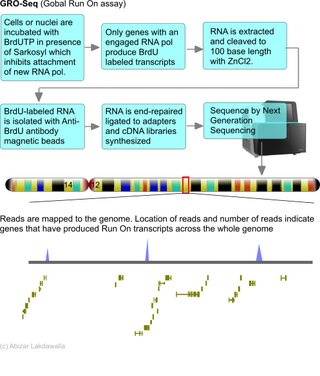
A nuclear run-on assay is conducted to identify the genes that are being transcribed at a certain time point. Approximately one million cell nuclei are isolated and incubated with labeled nucleotides, and genes in the process of being transcribed are detected by hybridization of extracted RNA to gene specific probes on a blot. Garcia-Martinez et al. (2004) developed a protocol for the yeast S. cerevisiae that allows for the calculation of transcription rates (TRs) for all yeast genes to estimate mRNA stabilities for all yeast mRNAs.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.

RIP-chip is a molecular biology technique which combines RNA immunoprecipitation with a microarray. The purpose of this technique is to identify which RNA sequences interact with a particular RNA binding protein of interest in vivo. It can also be used to determine relative levels of gene expression, to identify subsets of RNAs which may be co-regulated, or to identify RNAs that may have related functions. This technique provides insight into the post-transcriptional gene regulation which occurs between RNA and RNA binding proteins.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.

Chromatin immunoprecipitation (ChIP) is a type of immunoprecipitation experimental technique used to investigate the interaction between proteins and DNA in the cell. It aims to determine whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites, and possibly define cistromes. ChIP also aims to determine the specific location in the genome that various histone modifications are associated with, indicating the target of the histone modifiers. ChIP is crucial for the advancements in the field of epigenomics and learning more about epigenetic phenomena.
Peak calling is a computational method used to identify areas in a genome that have been enriched with aligned reads as a consequence of performing a ChIP-sequencing or MeDIP-seq experiment. These areas are those where a protein interacts with DNA. When the protein is a transcription factor, the enriched area is its transcription factor binding site (TFBS). Popular software programs include MACS. Wilbanks and colleagues is a survey of the ChIP-seq peak callers, and Bailey et al. is a description of practical guidelines for peak calling in ChIP-seq data.

ChIP-exo is a chromatin immunoprecipitation based method for mapping the locations at which a protein of interest binds to the genome. It is a modification of the ChIP-seq protocol, improving the resolution of binding sites from hundreds of base pairs to almost one base pair. It employs the use of exonucleases to degrade strands of the protein-bound DNA in the 5'-3' direction to within a small number of nucleotides of the protein binding site. The nucleotides of the exonuclease-treated ends are determined using some combination of DNA sequencing, microarrays, and PCR. These sequences are then mapped to the genome to identify the locations on the genome at which the protein binds.
Single-cell sequencing examines the sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
DRIP-seq (DRIP-sequencing) is a technology for genome-wide profiling of a type of DNA-RNA hybrid called an "R-loop". DRIP-seq utilizes a sequence-independent but structure-specific antibody for DNA-RNA immunoprecipitation (DRIP) to capture R-loops for massively parallel DNA sequencing.
ATAC-seq is a technique used in molecular biology to assess genome-wide chromatin accessibility. In 2013, the technique was first described as an alternative advanced method for MNase-seq, FAIRE-Seq and DNase-Seq. ATAC-seq is a faster and more sensitive analysis of the epigenome than DNase-seq or MNase-seq.

Chuan He is a Chinese-American chemical biologist. He currently serves as the John T. Wilson Distinguished Service Professor at the University of Chicago, and an Investigator of the Howard Hughes Medical Institute. He is best known for his work in discovering and deciphering reversible RNA methylation in post-transcriptional gene expression regulation.

In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.

Single cell epigenomics is the study of epigenomics in individual cells by single cell sequencing. Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.
H3K36ac is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the acetylation at the 36th lysine residue of the histone H3 protein.
ChIL sequencing (ChIL-seq), also known as Chromatin Integration Labeling sequencing, is a method used to analyze protein interactions with DNA. ChIL-sequencing combines antibody-targeted controlled cleavage by Tn5 transposase with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that ChIL-Sequencing does not. ChIL-Seq is a precise technique that reduces sample loss could be applied to single-cells.

MNase-seq, short for micrococcal nuclease digestion with deep sequencing, is a molecular biological technique that was first pioneered in 2006 to measure nucleosome occupancy in the C. elegans genome, and was subsequently applied to the human genome in 2008. Though, the term ‘MNase-seq’ had not been coined until a year later, in 2009. Briefly, this technique relies on the use of the non-specific endo-exonuclease micrococcal nuclease, an enzyme derived from the bacteria Staphylococcus aureus, to bind and cleave protein-unbound regions of DNA on chromatin. DNA bound to histones or other chromatin-bound proteins may remain undigested. The uncut DNA is then purified from the proteins and sequenced through one or more of the various Next-Generation sequencing methods.
References
- ↑ Meyer, Kate D.; Saletore, Yogesh; Zumbo, Paul; Elemento, Olivier; Mason, Christopher E.; Jaffrey, Samie R. (31 May 2012). "Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons". Cell. 149 (7): 1635–1646. doi:10.1016/j.cell.2012.05.003. PMC 3383396 . PMID 22608085.
- ↑ Dominissini, Dan; Moshitch-Moshkovitz, Sharon; Schwartz, Schraga; Salmon-Divon, Mali; Ungar, Lior; Osenberg, Sivan; Cesarkas, Karen; Jacob-Hirsch, Jasmine; Amariglio, Ninette; Kupiec, Martin; Sorek, Rotem; Rechavi, Gideon (28 April 2012). "Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq". Nature. 485 (7397): 201–206. Bibcode:2012Natur.485..201D. doi:10.1038/nature11112. PMID 22575960. S2CID 3517716.
- ↑ Delatte, Benjamin (2016). "Transcriptome-wide distribution and function of RNA hydroxymethylcytosine". Science. 351 (6270): 282–285. Bibcode:2016Sci...351..282D. doi: 10.1126/science.aac5253 . PMID 26816380.