
The Biopython project is an open-source collection of non-commercial Python tools for computational biology and bioinformatics, created by an international association of developers. It contains classes to represent biological sequences and sequence annotations, and it is able to read and write to a variety of file formats. It also allows for a programmatic means of accessing online databases of biological information, such as those at NCBI. Separate modules extend Biopython's capabilities to sequence alignment, protein structure, population genetics, phylogenetics, sequence motifs, and machine learning. Biopython is one of a number of Bio* projects designed to reduce code duplication in computational biology.

The Encyclopedia of DNA Elements (ENCODE) is a public research project which aims to identify functional elements in the human genome.

Google Analytics is a web analytics service offered by Google that tracks and reports website traffic, currently as a platform inside the Google Marketing Platform brand. Google launched the service in November 2005 after acquiring Urchin.

CLC bio was a bioinformatics software company headquartered in Aarhus, Denmark, and with offices in Cambridge, Massachusetts, Tokyo, Taipei and Delhi. CLC bio's software has more than 250,000 users in more than 100 countries around the globe. CLC bio was acquired by QIAGEN in 2013, and continues to exist as a part of QIAGEN Digital Insights, the bioinformatics research and development division of QIAGEN.
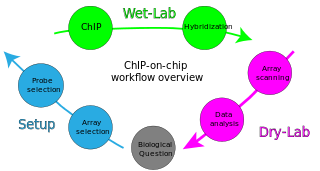
ChIP-on-chip is a technology that combines chromatin immunoprecipitation ('ChIP') with DNA microarray ("chip"). Like regular ChIP, ChIP-on-chip is used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis. Whole-genome analysis can be performed to determine the locations of binding sites for almost any protein of interest. As the name of the technique suggests, such proteins are generally those operating in the context of chromatin. The most prominent representatives of this class are transcription factors, replication-related proteins, like origin recognition complex protein (ORC), histones, their variants, and histone modifications.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.

UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.
Xenbase is a Model Organism Database (MOD), providing informatics resources, as well as genomic and biological data on Xenopus frogs. Xenbase has been available since 1999, and covers both X. laevis and X. tropicalis Xenopus varieties. As of 2013 all of its services are running on virtual machines in a private cloud environment, making it one of the first MODs to do so. Other than hosting genomics data and tools, Xenbase supports the Xenopus research community though profiles for researchers and laboratories, and job and events postings.

Integrated Genome Browser (IGB) is an open-source genome browser, a visualization tool used to observe biologically-interesting patterns in genomic data sets, including sequence data, gene models, alignments, and data from DNA microarrays.
The UCSC Genome Browser is an online and downloadable genome browser hosted by the University of California, Santa Cruz (UCSC). It is an interactive website offering access to genome sequence data from a variety of vertebrate and invertebrate species and major model organisms, integrated with a large collection of aligned annotations. The Browser is a graphical viewer optimized to support fast interactive performance and is an open-source, web-based tool suite built on top of a MySQL database for rapid visualization, examination, and querying of the data at many levels. The Genome Browser Database, browsing tools, downloadable data files, and documentation can all be found on the UCSC Genome Bioinformatics website.
Chromatin Interaction Analysis by Paired-End Tag Sequencing is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.
PATRIC is a bacterial bioinformatic website from the Bioinformatics Resource Center. It is an information system integrating databases with various types of data about bacterial pathogens together with analysis tools. Freely available, it is designed to support the biomedical research community's work on bacterial infectious diseases via these integrations of various pieces of pathogen information.
FAIRE-Seq is a method in molecular biology used for determining the sequences of DNA regions in the genome associated with regulatory activity. The technique was developed in the laboratory of Jason D. Lieb at the University of North Carolina, Chapel Hill. In contrast to DNase-Seq, the FAIRE-Seq protocol doesn't require the permeabilization of cells or isolation of nuclei, and can analyse any cell type. In a study of seven diverse human cell types, DNase-seq and FAIRE-seq produced strong cross-validation, with each cell type having 1-2% of the human genome as open chromatin.
DNAnexus is an American company that provides a cloud-based data analysis and management platform for DNA sequence data. It is based in Mountain View, California, and was founded in 2009 by Stanford University professors Serafim Batzoglou and Arend Sidow and Stanford computer scientist Andreas Sundquist.
The MEME suite is a collection of tools for the discovery and analysis of sequence motifs.
geWorkbench is an open-source software platform for integrated genomic data analysis. It is a desktop application written in the programming language Java. geWorkbench uses a component architecture. As of 2016, there are more than 70 plug-ins available, providing for the visualization and analysis of gene expression, sequence, and structure data.
Echinobase is a Model Organism Database (MOD). It supports the international research community by providing a centralized, integrated web based resource to access the diverse and rich, functional genomics data of echinoderm evolution, development and gene regulatory networks.
CUT&RUN sequencing, also known as cleavage under targets and release using nuclease, is a method used to analyze protein interactions with DNA. CUT&RUN sequencing combines antibody-targeted controlled cleavage by micrococcal nuclease with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN sequencing does not.