
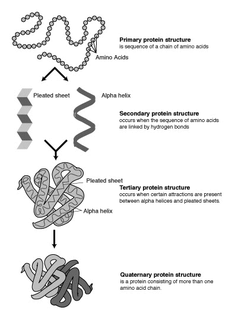
Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
Protein threading, also known as fold recognition, is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as it is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB), whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been seen that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
The global distance test (GDT), also written as GDT_TS to represent "total score", is a measure of similarity between two protein structures with known amino acid correspondences but different tertiary structures. It is most commonly used to compare the results of protein structure prediction to the experimentally determined structure as measured by X-ray crystallography, protein NMR, or, increasingly, cryoelectron microscopy. The metric was developed by Adam Zemla at Lawrence Livermore National Laboratory and originally implemented in the Local-Global Alignment (LGA) program. It is intended as a more accurate measurement than the common root-mean-square deviation (RMSD) metric - which is sensitive to outlier regions created, for example, by poor modeling of individual loop regions in a structure that is otherwise reasonably accurate. The conventional GDT_TS score is computed over the alpha carbon atoms and is reported as a percentage, ranging from 0 to 100. In general, the higher the GDT_TS score, the more closely a model approximates a given reference structure.
Modeller, often stylized as MODELLER, is a computer program used for homology modeling to produce models of protein tertiary structures and quaternary structures (rarer). It implements a method inspired by nuclear magnetic resonance spectroscopy of proteins, termed satisfaction of spatial restraints, by which a set of geometrical criteria are used to create a probability density function for the location of each atom in the protein. The method relies on an input sequence alignment between the target amino acid sequence to be modeled and a template protein which structure has been solved.
Loop modeling is a problem in protein structure prediction requiring the prediction of the conformations of loop regions in proteins with or without the use of a structural template. Computer programs that solve these problems have been used to research a broad range of scientific topics from ADP to breast cancer. Because protein function is determined by its shape and the physiochemical properties of its exposed surface, it is important to create an accurate model for protein/ligand interaction studies. The problem arises often in homology modeling, where the tertiary structure of an amino acid sequence is predicted based on a sequence alignment to a template, or a second sequence whose structure is known. Because loops have highly variable sequences even within a given structural motif or protein fold, they often correspond to unaligned regions in sequence alignments; they also tend to be located at the solvent-exposed surface of globular proteins and thus are more conformationally flexible. Consequently, they often cannot be modeled using standard homology modeling techniques. More constrained versions of loop modeling are also used in the data fitting stages of solving a protein structure by X-ray crystallography, because loops can correspond to regions of low electron density and are therefore difficult to resolve.
Anders Krogh is a bioinformatician at the University of Copenhagen, where he leads the university's bioinformatics center. He is known for his pioneering work on the use of hidden Markov models in bioinformatics, and is co-author of a widely used textbook in bioinformatics. In addition, he also co-authored one of the early textbooks on neural networks. His current research interests include promoter analysis, non-coding RNA, gene prediction and protein structure prediction.

RAPTOR is protein threading software used for protein structure prediction. It has been replaced by RaptorX, which is much more accurate than RAPTOR.
FoldX is a protein design algorithm that uses an empirical force field. It can determine the energetic effect of point mutations as well as the interaction energy of protein complexes. FoldX can mutate protein and DNA side chains using a probability-based rotamer library, while exploring alternative conformations of the surrounding side chains.
Protein backbone fragment libraries have been used successfully in a variety of structural biology applications, including homology modeling, de novo structure prediction, and structure determination. By reducing the complexity of the search space, these fragment libraries enable more rapid search of conformational space, leading to more efficient and accurate models.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.
PredictProtein (PP) is an automatic service that searches up-to-date public sequence databases, creates alignments, and predicts aspects of protein structure and function. Users send a protein sequence and receive a single file with results from database comparisons and prediction methods. PP went online in 1992 at the European Molecular Biology Laboratory; since 1999 it has operated from Columbia University and in 2009 it moved to the Technische Universität München. Although many servers have implemented particular aspects, PP remains the most widely used public server for structure prediction: over 1.5 million requests from users in 104 countries have been handled; over 13000 users submitted 10 or more different queries. PP web pages are mirrored in 17 countries on 4 continents. The system is optimized to meet the demands of experimentalists not experienced in bioinformatics. This implied that we focused on incorporating only high-quality methods, and tried to collate results omitting less reliable or less important ones.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.

In biochemistry, a backbone-dependent rotamer library provides the frequencies, mean dihedral angles, and standard deviations of the discrete conformations of the amino acid side chains in proteins as a function of the backbone dihedral angles φ and ψ of the Ramachandran map. By contrast, backbone-independent rotamer libraries express the frequencies and mean dihedral angles for all side chains in proteins, regardless of the backbone conformation of each residue type. Backbone-dependent rotamer libraries have been shown to have significant advantages over backbone-independent rotamer libraries, principally when used as an energy term, by speeding up search times of side-chain packing algorithms used in protein structure prediction and protein design.