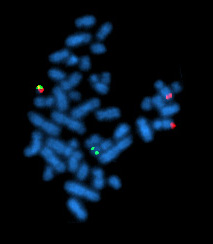
Image: example of karyotyping showing a total of 46 chromosomes in the genome.
Molecular cytogenetics combines two disciplines, molecular biology and cytogenetics, and involves the analysis of chromosome structure to help distinguish normal and cancer-causing cells. Human cytogenetics began in 1956 when it was discovered that normal human cells contain 46 chromosomes. However, the first microscopic observations of chromosomes were reported by Arnold, Flemming, and Hansemann in the late 1800s. Their work was ignored for decades until the actual chromosome number in humans was discovered as 46. In 1879, Arnold examined sarcoma and carcinoma cells having very large nuclei. Today, the study of molecular cytogenetics can be useful in diagnosing and treating various malignancies such as hematological malignancies, brain tumors, and other precursors of cancer. The field is overall focused on studying the evolution of chromosomes, more specifically the number, structure, function, and origin of chromosome abnormalities.[1][2] It includes a series of techniques referred to as fluorescence in situ hybridization, or FISH, in which DNA probes are labeled with different colored fluorescent tags to visualize one or more specific regions of the genome. Introduced in the 1980s, FISH uses probes with complementary base sequences to locate the presence or absence of the specific DNA regions. FISH can either be performed as a direct approach to metaphase chromosomes or interphase nuclei. Alternatively, an indirect approach can be taken in which the entire genome can be assessed for copy number changes using virtual karyotyping. Virtual karyotypes are generated from arrays made of thousands to millions of probes, and computational tools are used to recreate the genome in silico.
FISH images of chromosomes from dividing orangutan (left) and human (right) cells. Yellow probe shows 4 copies of a region in the orangutan genome and only 2 copies in human.
Fluorescence In Situ Hybridization maps out single copy or repetitive DNA sequences through localization labeling of specific nucleic acids. The technique utilizes different DNA probes labeled with fluorescent tags that bind to one or more specific regions of the genome.[3] It labels all individual chromosomes at every stage of cell division to display structural and numerical abnormalities that may arise throughout the cycle. This is done with a probe that can be locus specific, centromeric, telomeric, and whole-chromosomal. This technique is typically performed on interphase cells and paraffin block tissues. FISH maps out single copy or repetitive DNA sequences through localization labeling of specific nucleic acids. The technique utilizes different DNA probes labeled with fluorescent tags that bind to one or more specific regions of the genome. Signals from the fluorescent tags can be seen with microscopy, and mutations can be seen by comparing these signals to healthy cells. For this to work, DNA must be denatured using heat or chemicals to break the hydrogen bonds; this allows hybridization to occur once two samples are mixed. The fluorescent probes create new hydrogen bonds, thus repairing DNA with their complementary bases, which can be detected through microscopy. FISH allows one to visualize different parts of the chromosome at different stages of the cell cycle. FISH can either be performed as a direct approach to metaphase chromosomes or interphase nuclei. Alternatively, an indirect approach can be taken in which the entire genome can be assessed for copy number changes using virtual karyotyping. Virtual karyotypes are generated from microarrays made of thousands to millions of probes, and computational tools are used to recreate the genome in silico.[4]
Comparative genomic hybridization (CGH)
Comparative genomic hybridization (CGH), derived from FISH, is used to compare variations in copy number between a biological sample and a reference. CGH was originally developed to observe chromosomal aberrations in tumour cells. This method uses two genomes, a sample and a control, which are labeled fluorescently to distinguish them.[5] In CGH, DNA is isolated from a tumour sample and biotin is attached. Another labelling protein, digoxigenin, is attached to the reference DNA sample.[6] The labelled DNA samples are co-hybridized to probes during cell division, which is the most informative time for observing copy number variation.[7] CGH uses creates a map that shows the relative abundance of DNA and chromosome number. By comparing the fluorescence in a sample compared to a reference, CGH can point to gains or losses of chromosomal regions.[6][8] CGH differs from FISH because it does not require a specific target or previous knowledge of the genetic region being analyzed. CGH can also scan an entire genome relatively quickly for various chromosome imbalances, and this is helpful in patients with underlying genetic issues and when an official diagnosis is not known. This often occurs with hematological cancers.
Array comparative genomic hybridization (aCGH)
Array comparative genomic hybridization (aCGH) allows CGH to be performed without cell culture and isolation. Instead, it is performed on glass slides containing small DNA fragments.[9] Removing the cell culture and isolation step dramatically simplifies and expedites the process. Using similar principles to CGH, the sample DNA is isolated and fluorescently labelled, then co-hybridized to single stranded probes to generate signals. Thousands of these signals can be detected for at once, and this process is referred to as parallel screening.[10] Fluorescence ratios between the sample and reference signals are measured, representing the average difference between the amount of each. This will show if there is more or less sample DNA than is expected by reference.
Applications
A cell containing a rearrangement of the bcr/abl chromosomal regions (upper left red and green chromosome). This rearrangement is associated with chronic myelogenous leukemia, and was detected using FISH.
FISH chromosome in-situ hybridization allows the study cytogenetics in pre- and postnatal samples and is also widely used in cytogenetic testing for cancer. While cytogenetics is the study of chromosomes and their structure, cytogenetic testing involves the analysis of cells in the blood, tissue, bone marrow, or fluid to identify changes in chromosomes of an individual. This was often done through karyotyping, and is now done with FISH. This method is commonly used to detect chromosomal deletions or translocations often associated with cancer. FISH is also used for melanocytic lesions, distinguishing atypical melanocytic or malignant melanoma.[5]
Cancer cells often accumulate complex chromosomal structural changes such as loss, duplication, inversion or movement of a segment.[11] When using FISH, any changes to a chromosome will be made visible through discrepancies between fluorescent-labelled cancer chromosomes and healthy chromosomes.[11] The findings of these cytogenetic experiments can shed light on the genetic causes for the cancer and can locate potential therapeutic targets.[12]
Molecular cytogenetics can also be used as a diagnostic tool for congenital syndromes in which the underlying genetic causes of the disease are unknown.[13] Analysis of a patient's chromosome structure can reveal causative changes. New molecular biology methods developed in the past two decades such as next generation sequencing and RNA-seq have largely replaced molecular cytogenetics in diagnostics, but recently the use of derivatives of FISH such as multicolour FISH and multicolour banding (mBAND) has been growing in medical applications.[14]
Cancer projects
One of the current projects involving Molecular Cytogenetics involves genomic research on rare cancers, called the Cancer Genome Characterization Initiative (CGCI).[15] The CGCI is a group interested in describing the genetic abnormalities of some rare cancers, by employing advanced sequencing of genomes, exomes, and transcriptomes, which may ultimately play a role in cancer pathogenesis.[15] Currently, the CGCI has elucidated some previously undetermined genetic alterations in medulloblastoma and B-cell non-Hodgkin lymphoma. The next steps for the CGCI is to identify genomic alternations in HIV+ tumors and in Burkitt's Lymphoma.
1 2 Rao PH, Nandula SV, Murty VV (2007). "Molecular cytogenetic applications in analysis of the cancer genome". In Fisher PB (ed.). Cancer Genomics and Proteomics: Methods and Protocols. Methods in Molecular Biology. Vol.383. Humana Press. pp.165–85. doi:10.1007/978-1-59745-335-6_11. ISBN9781597453356. PMID18217685.
↑ Speicher MR, Carter NP (October 2005). "The new cytogenetics: blurring the boundaries with molecular biology". Nature Reviews Genetics. 6 (10): 782–92. doi:10.1038/nrg1692. PMID16145555. S2CID15023775.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.