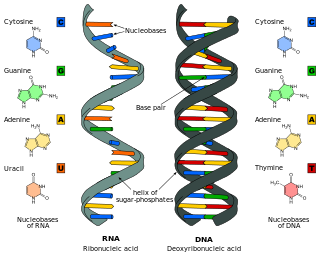
Nucleic acids are biopolymers, macromolecules, essential to all known forms of life. They are composed of nucleotides, which are the monomers made of three components: a 5-carbon sugar, a phosphate group and a nitrogenous base. The two main classes of nucleic acids are deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). If the sugar is ribose, the polymer is RNA; if the sugar is the ribose derivative deoxyribose, the polymer is DNA.
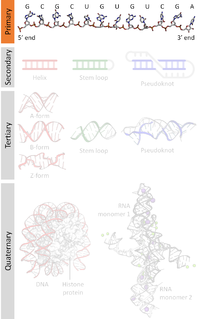
A nucleic acid sequence is a succession of bases signified by a series of a set of five different letters that indicate the order of nucleotides forming alleles within a DNA or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
In the field of bioinformatics, a sequence database is a type of biological database that is composed of a large collection of computerized ("digital") nucleic acid sequences, protein sequences, or other polymer sequences stored on a computer. The UniProt database is an example of a protein sequence database. As of 2013 it contained over 40 million sequences and is growing at an exponential rate. Historically, sequences were published in paper form, but as the number of sequences grew, this storage method became unsustainable.
In biology, a sequence motif is a nucleotide or amino-acid sequence pattern that is widespread and usually assumed to be related to biological function of the macromolecule. For example, an N-glycosylation site motif can be defined as Asn, followed by anything but Pro, followed by either Ser or Thr, followed by anything but Pro residue.
In bioinformatics and biochemistry, the FASTA format is a text-based format for representing either nucleotide sequences or amino acid (protein) sequences, in which nucleotides or amino acids are represented using single-letter codes. The format also allows for sequence names and comments to precede the sequences. The format originates from the FASTA software package, but has now become a near universal standard in the field of bioinformatics.
In statistics and related fields, a similarity measure or similarity function or similarity metric is a real-valued function that quantifies the similarity between two objects. Although no single definition of a similarity exists, usually such measures are in some sense the inverse of distance metrics: they take on large values for similar objects and either zero or a negative value for very dissimilar objects. Though, in more broad terms, a similarity function may also satisfy metric axioms.
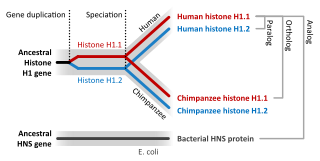
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

Ensembl genome database project is a scientific project at the European Bioinformatics Institute, which was launched in 1999 in response to the imminent completion of the Human Genome Project. Ensembl aims to provide a centralized resource for geneticists, molecular biologists and other researchers studying the genomes of our own species and other vertebrates and model organisms. Ensembl is one of several well known genome browsers for the retrieval of genomic information.
The European Bioinformatics Institute (EMBL-EBI) is an Intergovernmental Organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.

Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 34.0, was released in March 2021 and contains 19,179 families.
The ARB Project is a free software package for phylogenetic analysis of rRNA and other biological sequences such as amino acids. It allows an innovative and alternative execution of phylogeny. It also simplifies the import and assembly of genetic sequences from any organisms with the use of an automated aligner. This editing process can be depicted into two sub categories: "Primary Structure Editor" and "Secondary Structure Editor." Comprehensively, this allows the necessary make up for the "Phylogenetic Treeing." The software also allows the visualization of these biological sequences in which allows the user a more in depth experience and interaction. This is particularly necessary when comparing phylogeny data from various organisms.
Rfam is a database containing information about non-coding RNA (ncRNA) families and other structured RNA elements. It is an annotated, open access database originally developed at the Wellcome Trust Sanger Institute in collaboration with Janelia Farm, and currently hosted at the European Bioinformatics Institute. Rfam is designed to be similar to the Pfam database for annotating protein families.
Napp or NAPP may refer to:

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.
28S ribosomal RNA is the structural ribosomal RNA (rRNA) for the large subunit (LSU) of eukaryotic cytoplasmic ribosomes, and thus one of the basic components of all eukaryotic cells. It has a size of 25S in plants and 28S in mammals, hence the alias of 25S–28S rRNA.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.
In bioinformatics, the PANTHER classification system is a large curated biological database of gene/protein families and their functionally related subfamilies that can be used to classify and identify the function of gene products. PANTHER is part of the Gene Ontology Reference Genome Project designed to classify proteins and their genes for high-throughput analysis.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.