
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.

The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.
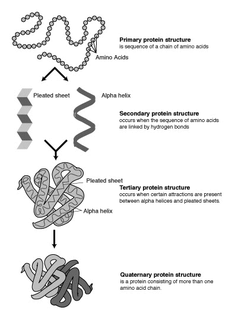
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.
The European Bioinformatics Institute (EMBL-EBI) is an Intergovernmental Organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.

Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 34.0, was released in March 2021 and contains 19,179 families.
HomoloGene, a tool of the United States National Center for Biotechnology Information (NCBI), is a system for automated detection of homologs among the annotated genes of several completely sequenced eukaryotic genomes.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
InterPro is a database of protein families, domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
Protein–protein interaction prediction is a field combining bioinformatics and structural biology in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding protein–protein interactions is important for the investigation of intracellular signaling pathways, modelling of protein complex structures and for gaining insights into various biochemical processes.
Stockholm format is a multiple sequence alignment format used by Pfam and Rfam to disseminate protein and RNA sequence alignments. The alignment editors Ralee , Belvu and Jalview support Stockholm format as do the probabilistic database search tools, Infernal and HMMER, and the phylogenetic analysis tool Xrate. Stockholm format files often have the filename extension .sto or .stk.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and Mac OS.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.
The Conserved Domain Database (CDD) is a database of well-annotated multiple sequence alignment models and derived database search models, for ancient domains and full-length proteins.
PhylomeDB is a public biological database for complete catalogs of gene phylogenies (phylomes). It allows users to interactively explore the evolutionary history of genes through the visualization of phylogenetic trees and multiple sequence alignments. Moreover, phylomeDB provides genome-wide orthology and paralogy predictions which are based on the analysis of the phylogenetic trees. The automated pipeline used to reconstruct trees aims at providing a high-quality phylogenetic analysis of different genomes, including Maximum Likelihood tree inference, alignment trimming and evolutionary model testing.
The Virus Pathogen Database and Analysis Resource (ViPR) is an integrative and comprehensive publicly available database and analysis resource to search, analyze, visualize, save and share data for viral pathogens in the U.S. National Institute of Allergy and Infectious Diseases (NIAID) Category A-C Priority Pathogen lists for biodefense research, and other viral pathogens causing emerging/reemerging infectious diseases. ViPR is one of the five Bioinformatics Resource Centers (BRC) funded by NIAID, a component of the National Institutes of Health (NIH), which is an agency of the United States Department of Health and Human Services.
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.