
The citric acid cycle (CAC)—also known as the Krebs cycle, Szent-Györgyi-Krebs cycle or the TCA cycle (tricarboxylic acid cycle)—is a series of chemical reactions to release stored energy through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins. The Krebs cycle is used by organisms that respire (as opposed to organisms that ferment) to generate energy, either by anaerobic respiration or aerobic respiration. In addition, the cycle provides precursors of certain amino acids, as well as the reducing agent NADH, that are used in numerous other reactions. Its central importance to many biochemical pathways suggests that it was one of the earliest components of metabolism. Even though it is branded as a 'cycle', it is not necessary for metabolites to follow only one specific route; at least three alternative segments of the citric acid cycle have been recognized.
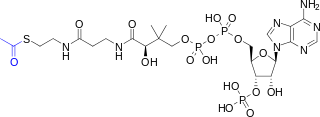
Acetyl-CoA is a molecule that participates in many biochemical reactions in protein, carbohydrate and lipid metabolism. Its main function is to deliver the acetyl group to the citric acid cycle to be oxidized for energy production. Coenzyme A consists of a β-mercaptoethylamine group linked to the vitamin pantothenic acid (B5) through an amide linkage and 3'-phosphorylated ADP. The acetyl group of acetyl-CoA is linked to the sulfhydryl substituent of the β-mercaptoethylamine group. This thioester linkage is a "high energy" bond, which is particularly reactive. Hydrolysis of the thioester bond is exergonic (−31.5 kJ/mol).
Gluconeogenesis (GNG) is a metabolic pathway that results in the generation of glucose from certain non-carbohydrate carbon substrates. It is a ubiquitous process, present in plants, animals, fungi, bacteria, and other microorganisms. In vertebrates, gluconeogenesis occurs mainly in the liver and, to a lesser extent, in the cortex of the kidneys. It is one of two primary mechanisms – the other being degradation of glycogen (glycogenolysis) – used by humans and many other animals to maintain blood sugar levels, avoiding low levels (hypoglycemia). In ruminants, because dietary carbohydrates tend to be metabolized by rumen organisms, gluconeogenesis occurs regardless of fasting, low-carbohydrate diets, exercise, etc. In many other animals, the process occurs during periods of fasting, starvation, low-carbohydrate diets, or intense exercise.
Lactic acidosis is a medical condition characterized by a build-up of lactate in the body, with formation of an excessively low pH in the bloodstream. It is a form of metabolic acidosis, in which excessive acid accumulates due to a problem with the body's oxidative metabolism.

Tumor hypoxia is the situation where tumor cells have been deprived of oxygen. As a tumor grows, it rapidly outgrows its blood supply, leaving portions of the tumor with regions where the oxygen concentration is significantly lower than in healthy tissues. Hypoxic microenvironements in solid tumors are a result of available oxygen being consumed within 70 to 150 μm of tumour vasculature by rapidly proliferating tumor cells thus limiting the amount of oxygen available to diffuse further into the tumor tissue. In order to support continuous growth and proliferation in challenging hypoxic environments, cancer cells are found to alter their metabolism. Furthermore, hypoxia is known to change cell behavior and is associated with extracellular matrix remodeling and increased migratory and metastatic behavior.

The Cori cycle, named after its discoverers, Carl Ferdinand Cori and Gerty Cori, is a metabolic pathway in which lactate, produced by anaerobic glycolysis in muscles, is transported to the liver and converted to glucose, which then returns to the muscles and is cyclically metabolized back to lactate.
In oncology, the Warburg effect is the observation that most cancer cells produce energy predominantly not through the 'usual' citric acid cycle and oxidative phosphorylation in the mitochondria as observed in normal cells, but through a less efficient process of 'aerobic glycolysis' consisting of a high level of glucose uptake and glycolysis followed by lactic acid fermentation taking place in the cytosol, not the mitochondria, even in the presence of abundant oxygen. This observation was first published by Otto Heinrich Warburg, who was awarded the 1931 Nobel Prize in Physiology for his "discovery of the nature and mode of action of the respiratory enzyme". The precise mechanism and therapeutic implications of the Warburg effect, however, remain unclear.
Pyruvate dehydrogenase lipoamide kinase isozyme 1, mitochondrial is an enzyme that in humans is encoded by the PDK1 gene. It codes for an isozyme of pyruvate dehydrogenase kinase (PDK).
Pyruvate dehydrogenase deficiency is a rare neurodegenerative disorder associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC). The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria. The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.
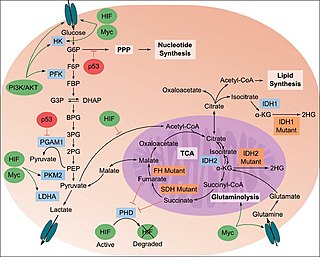
The study of the tumor metabolism, also known as tumor metabolome describes the different characteristic metabolic changes in tumor cells. The characteristic attributes of the tumor metabolome are high glycolytic enzyme activities, the expression of the pyruvate kinase isoenzyme type M2, increased channeling of glucose carbons into synthetic processes, such as nucleic acid, amino acid and phospholipid synthesis, a high rate of pyrimidine and purine de novo synthesis, a low ratio of Adenosine triphosphate and Guanosine triphosphate to Cytidine triphosphate and Uridine triphosphate, low Adenosine monophosphate levels, high glutaminolytic capacities, release of immunosuppressive substances and dependency on methionine.

Dihydrolipoyl transacetylase is an enzyme component of the multienzyme pyruvate dehydrogenase complex. The pyruvate dehydrogenase complex is responsible for the pyruvate decarboxylation step that links glycolysis to the citric acid cycle. This involves the transformation of pyruvate from glycolysis into acetyl-CoA which is then used in the citric acid cycle to carry out cellular respiration.

Pyruvate dehydrogenase kinase is a kinase enzyme which acts to inactivate the enzyme pyruvate dehydrogenase by phosphorylating it using ATP.

Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial is an enzyme that in humans is encoded by the PDHA1 gene.The pyruvate dehydrogenase complex is a nuclear-encoded mitochondrial matrix multienzyme complex that provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle by catalyzing the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of 3 enzymes: E1 (PDHA1); dihydrolipoyl transacetylase (DLAT) ; and dihydrolipoyl dehydrogenase (DLD). The E1 enzyme is a heterotetramer of 2 alpha and 2 beta subunits. The E1-alpha subunit contains the E1 active site and plays a key role in the function of the PDH complex.

Lactate dehydrogenase (LDH or LD) is an enzyme found in nearly all living cells. LDH catalyzes the conversion of pyruvate to lactate and back, as it converts NAD+ to NADH and back. A dehydrogenase is an enzyme that transfers a hydride from one molecule to another.
Pyruvate dehydrogenase lipoamide kinase isozyme 4, mitochondrial (PDK4) is an enzyme that in humans is encoded by the PDK4 gene. It codes for an isozyme of pyruvate dehydrogenase kinase.

Pyruvate dehydrogenase kinase isoform 2 (PDK2) also known as pyruvate dehydrogenase lipoamide kinase isozyme 2, mitochondrial is an enzyme that in humans is encoded by the PDK2 gene. PDK2 is an isozyme of pyruvate dehydrogenase kinase.

Pyruvate dehydrogenase lipoamide kinase isozyme 3, mitochondrial is an enzyme that in humans is encoded by the PDK3 gene. It codes for an isozyme of pyruvate dehydrogenase kinase.The pyruvate dehydrogenase (PDH) complex is a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO2. It provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle, and thus is one of the major enzymes responsible for the regulation of glucose metabolism. The enzymatic activity of PDH is regulated by a phosphorylation/dephosphorylation cycle, and phosphorylation results in inactivation of PDH. The protein encoded by this gene is one of the four pyruvate dehydrogenase kinases that inhibits the PDH complex by phosphorylation of the E1 alpha subunit. This gene is predominantly expressed in the heart and skeletal muscles. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.

Pyruvate dehydrogenase (lipoamide) beta, also known as pyruvate dehydrogenase E1 component subunit beta, mitochondrial or PDHE1-B is an enzyme that in humans is encoded by the PDHB gene. The pyruvate dehydrogenase (PDH) complex is a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO2, and provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle. The PDH complex is composed of multiple copies of three enzymatic components: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2) and lipoamide dehydrogenase (E3). The E1 enzyme is a heterotetramer of two alpha and two beta subunits. This gene encodes the E1 beta subunit. Mutations in this gene are associated with pyruvate dehydrogenase E1-beta deficiency.
The lactate shuttle hypothesis describes the movement of lactate intracellularly and intercellularly. The hypothesis is based on the observation that lactate is formed and utilized continuously in diverse cells under both anaerobic and aerobic conditions. Further, lactate produced at sites with high rates of glycolysis and glycogenolysis can be shuttled to adjacent or remote sites including heart or skeletal muscles where the lactate can be used as a gluconeogenic precursor or substrate for oxidation. The hypothesis was proposed by professor George Brooks of the University of California at Berkeley.