
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
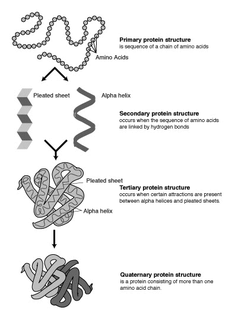
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Biological databases are libraries of biological sciences, collected from scientific experiments, published literature, high-throughput experiment technology, and computational analysis. They contain information from research areas including genomics, proteomics, metabolomics, microarray gene expression, and phylogenetics. Information contained in biological databases includes gene function, structure, localization, clinical effects of mutations as well as similarities of biological sequences and structures.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
A sequence profiling tool in bioinformatics is a type of software that presents information related to a genetic sequence, gene name, or keyword input. Such tools generally take a query such as a DNA, RNA, or protein sequence or ‘keyword’ and search one or more databases for information related to that sequence. Summaries and aggregate results are provided in standardized format describing the information that would otherwise have required visits to many smaller sites or direct literature searches to compile. Many sequence profiling tools are software portals or gateways that simplify the process of finding information about a query in the large and growing number of bioinformatics databases. The access to these kinds of tools is either web based or locally downloadable executables.

Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 34.0, was released in March 2021 and contains 19,179 families.
InterPro is a database of protein families, domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
Peroxiredoxins are a ubiquitous family of antioxidant enzymes that also control cytokine-induced peroxide levels and thereby mediate signal transduction in mammalian cells. The family members in humans are PRDX1, PRDX2, PRDX3, PRDX4, PRDX5, and PRDX6. The physiological importance of peroxiredoxins is illustrated by their relative abundance.
Rfam is a database containing information about non-coding RNA (ncRNA) families and other structured RNA elements. It is an annotated, open access database originally developed at the Wellcome Trust Sanger Institute in collaboration with Janelia Farm, and currently hosted at the European Bioinformatics Institute. Rfam is designed to be similar to the Pfam database for annotating protein families.

The PeroxiBase database has been created at the University of Geneva (Switzerland) at the end of 2003, by two plant biologists specialised in the study of plant peroxidases. It was first limited to class III peroxidases and was then expanded to include all possible haem and non-haem peroxidase protein sequences. Many researchers and bioinformaticians from the University of Geneva joined their efforts to develop the database and rapidly increase the number of peroxidase sequences. Since 2005, the database accepts external contributions, which are verified by PeroxiBase curators. The majority of haem and non-haem peroxidase sequences can now be found in the PeroxiBase.
In molecular biology, the PRINTS database is a collection of so-called "fingerprints": it provides both a detailed annotation resource for protein families, and a diagnostic tool for newly determined sequences. A fingerprint is a group of conserved motifs taken from a multiple sequence alignment - together, the motifs form a characteristic signature for the aligned protein family. The motifs themselves are not necessarily contiguous in sequence, but may come together in 3D space to define molecular binding sites or interaction surfaces. The particular diagnostic strength of fingerprints lies in their ability to distinguish sequence differences at the clan, superfamily, family and subfamily levels. This allows fine-grained functional diagnoses of uncharacterised sequences, allowing, for example, discrimination between family members on the basis of the ligands they bind or the proteins with which they interact, and highlighting potential oligomerisation or allosteric sites.

Peroxiredoxin-2 is a protein that in humans is encoded by the PRDX2 gene.
Peroxiredoxin-5 (PRDX5), mitochondrial is a protein that in humans is encoded by the PRDX5 gene, located on chromosome 11.
Stockholm format is a multiple sequence alignment format used by Pfam and Rfam to disseminate protein and RNA sequence alignments. The alignment editors Ralee , Belvu and Jalview support Stockholm format as do the probabilistic database search tools, Infernal and HMMER, and the phylogenetic analysis tool Xrate. Stockholm format files often have the filename extension .sto or .stk.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and Mac OS.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.
In bioinformatics, the PANTHER classification system is a large curated biological database of gene/protein families and their functionally related subfamilies that can be used to classify and identify the function of gene products. PANTHER is part of the Gene Ontology Reference Genome Project designed to classify proteins and their genes for high-throughput analysis.
Jacquelyn S. (Jacque) Fetrow is a computational biologist, college administrator, and the 15th president of Albright College. Previously, she served as Provost, Vice President of Academic Affairs, and Professor of Chemistry at the University of Richmond, in Richmond, Virginia. Prior to that appointment, she served as Dean of the College at Wake Forest University in Winston-Salem, North Carolina. She also co-founded a company, GeneFormatics, for which she served as Director and Chief Scientific Officer for four years.