Aromatic compounds are those chemical compounds that contain one or more rings with pi electrons delocalized all the way around them. In contrast to compounds that exhibit aromaticity, aliphatic compounds lack this delocalization. The term "aromatic" was assigned before the physical mechanism determining aromaticity was discovered, and referred simply to the fact that many such compounds have a sweet or pleasant odour; however, not all aromatic compounds have a sweet odour, and not all compounds with a sweet odour are aromatic. Aromatic hydrocarbons, or arenes, are aromatic organic compounds containing solely carbon and hydrogen atoms. The configuration of six carbon atoms in aromatic compounds is called a "benzene ring", after the simple aromatic compound benzene, or a phenyl group when part of a larger compound.

Organolithium reagents are organometallic compounds that contain carbon – lithium bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C-Li bond is highly ionic. Owing to the polar nature of the C-Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
Arynes or benzynes are highly reactive species derived from an aromatic ring by removal of two substituents. The most common arynes are ortho but meta- and para-arynes are also known. o-Arynes are examples of strained alkynes.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
A cascade reaction, also known as a domino reaction or tandem reaction, is a chemical process that comprises at least two consecutive reactions such that each subsequent reaction occurs only in virtue of the chemical functionality formed in the previous step. In cascade reactions, isolation of intermediates is not required, as each reaction composing the sequence occurs spontaneously. In the strictest definition of the term, the reaction conditions do not change among the consecutive steps of a cascade and no new reagents are added after the initial step. By contrast, one-pot procedures similarly allow at least two reactions to be carried out consecutively without any isolation of intermediates, but do not preclude the addition of new reagents or the change of conditions after the first reaction. Thus, any cascade reaction is also a one-pot procedure, while the reverse does not hold true. Although often composed solely of intramolecular transformations, cascade reactions can also occur intermolecularly, in which case they also fall under the category of multicomponent reactions.

A formylation reaction in organic chemistry refers to organic reactions in which an organic compound is functionalized with a formyl group (-CH=O). The reaction is a route to aldehydes (C-CH=O), formamides (N-CH=O), and formate esters (O-CH=O). A reagent that delivers the formyl group is called a formylating agent. A particularly important formylation process is hydroformylation which converts alkenes to the homologated aldehyde. The conversion of benzene to benzaldehyde is the basis of the Gattermann–Koch reaction:
The Barton–Kellogg reaction is a coupling reaction between a diazo compound and a thioketone, giving an alkene by way of an episulfide intermediate. The Barton–Kellogg reaction is also known as Barton–Kellogg olefination and Barton olefin synthesis.

The Dakin oxidation is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde or ketone reacts with hydrogen peroxide in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidized, and the hydrogen peroxide is reduced.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.

Aziridines are organic compounds containing the aziridine functional group, a three-membered heterocycle with one amine (-NR-) and two methylene bridges. The parent compound is aziridine, with molecular formula C
2H
4NH. Several drugs feature aziridine rings, including mitomycin C, porfiromycin, and azinomycin B (carzinophilin).
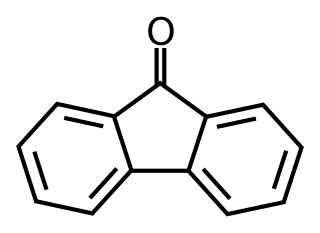
Fluorenone is an aromatic organic compound with the chemical formula C13H8O. It is used to make antimalaria drugs. It can be synthesised from fluorene with the addition of glacial acetic acid and sodium hypochlorite solution, undergoing an oxidation reaction. It is bright fluorescent yellow in color and is a solid at room temperature.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.
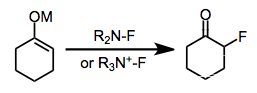
Electrophilic fluorination is the combination of a carbon-centered nucleophile with an electrophilic source of fluorine to afford organofluorine compounds. Although elemental fluorine and reagents incorporating an oxygen-fluorine bond can be used for this purpose, they have largely been replaced by reagents containing a nitrogen-fluorine bond.

Stilbene photocyclization is the coupling of two aromatic carbons in stilbenes upon ultraviolet irradiation. The reaction can be used to form polycyclic aromatic hydrocarbons and heteroaromatics.
Carbonyl oxidation with hypervalent iodine reagents involves the functionalization of the α position of carbonyl compounds through the intermediacy of a hypervalent iodine(III) enolate species. This electrophilic intermediate may be attacked by a variety of nucleophiles or undergo rearrangement or elimination.
The Payne rearrangement is the isomerization, under basic conditions, of 2,3-epoxy alcohols to isomeric 2,3-epoxy alcohols with inversion of configuration. Aza- and thia-Payne rearrangements of aziridines and thiiraniums, respectively, are also known.
Benzylic activation and stereocontrol in tricarbonyl(arene)chromium complexes refers to the enhanced rates and stereoselectivities of reactions at the benzylic position of aromatic rings complexed to chromium(0) relative to uncomplexed arenes. Complexation of an aromatic ring to chromium stabilizes both anions and cations at the benzylic position and provides a steric blocking element for diastereoselective functionalization of the benzylic position. A large number of stereoselective methods for benzylic and homobenzylic functionalization have been developed based on this property.
Reactions of organocopper reagents involve species containing copper-carbon bonds acting as nucleophiles in the presence of organic electrophiles. Organocopper reagents are now commonly used in organic synthesis as mild, selective nucleophiles for substitution and conjugate addition reactions.
Trifluoromethylation in organic chemistry describes any organic reaction that introduces a trifluoromethyl group in an organic compound. Trifluoromethylated compounds are of some importance in pharmaceutical industry and agrochemicals. Several notable pharmaceutical compounds have a trifluoromethyl group incorporated: fluoxetine, mefloquine, Leflunomide, nulitamide, dutasteride, bicalutamide, aprepitant, celecoxib, fipronil, fluazinam, penthiopyrad, picoxystrobin, fluridone, norflurazon, sorafenib and triflurazin. A relevant agrochemical is trifluralin. The development of synthetic methods for adding trifluoromethyl groups to chemical compounds is actively pursued in academic research.
In organic chemistry, the hexadehydro-Diels–Alder (HDDA) reaction is an organic chemical reaction between a diyne and an alkyne to form a reactive benzyne species, via a [4+2] cycloaddition reaction. This benzyne intermediate then reacts with a suitable trapping agent to form a substituted aromatic product. This reaction is a derivative of the established Diels–Alder reaction and proceeds via a similar [4+2] cycloaddition mechanism. The HDDA reaction is particularly effective for forming heavily functionalized aromatic systems and multiple ring systems in one synthetic step.













