Related Research Articles
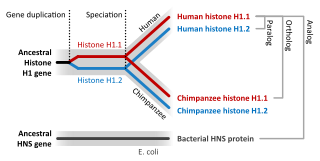
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
The European Bioinformatics Institute (EMBL-EBI) is an Intergovernmental Organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.
Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 34.0, was released in March 2021 and contains 19,179 families.
InterPro is a database of protein families, domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
Protein–protein interaction prediction is a field combining bioinformatics and structural biology in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding protein–protein interactions is important for the investigation of intracellular signaling pathways, modelling of protein complex structures and for gaining insights into various biochemical processes.

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.

In molecular biology, STRING is a biological database and web resource of known and predicted protein–protein interactions.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.
The UCSC Genome Browser is an online and downloadable genome browser hosted by the University of California, Santa Cruz (UCSC). It is an interactive website offering access to genome sequence data from a variety of vertebrate and invertebrate species and major model organisms, integrated with a large collection of aligned annotations. The Browser is a graphical viewer optimized to support fast interactive performance and is an open-source, web-based tool suite built on top of a MySQL database for rapid visualization, examination, and querying of the data at many levels. The Genome Browser Database, browsing tools, downloadable data files, and documentation can all be found on the UCSC Genome Bioinformatics website.
OrthoDB presents a catalog of orthologous protein-coding genes across vertebrates, arthropods, fungi, plants, and bacteria. Orthology refers to the last common ancestor of the species under consideration, and thus OrthoDB explicitly delineates orthologs at each major radiation along the species phylogeny. The database of orthologs presents available protein descriptors, together with Gene Ontology and InterPro attributes, which serve to provide general descriptive annotations of the orthologous groups, and facilitate comprehensive orthology database querying. OrthoDB also provides computed evolutionary traits of orthologs, such as gene duplicability and loss profiles, divergence rates, sibling groups, and gene intron-exon architectures.
OMA is a database of orthologs extracted from available complete genomes. The orthology predictions of OMA are available in several forms:
Blast2GO, first published in 2005, is a bioinformatics software tool for the automatic, high-throughput functional annotation of novel sequence data. It makes use of the BLAST algorithm to identify similar sequences to then transfers existing functional annotation from yet characterised sequences to the novel one. The functional information is represented via the Gene Ontology (GO), a controlled vocabulary of functional attributes. The Gene Ontology, or GO, is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species.
In bioinformatics, the PANTHER classification system is a large curated biological database of gene/protein families and their functionally related subfamilies that can be used to classify and identify the function of gene products. PANTHER is part of the Gene Ontology Reference Genome Project designed to classify proteins and their genes for high-throughput analysis.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.
In molecular phylogenetics, relationships among individuals are determined using character traits, such as DNA, RNA or protein, which may be obtained using a variety of sequencing technologies. High-throughput next-generation sequencing has become a popular technique in transcriptomics, which represent a snapshot of gene expression. In eukaryotes, making phylogenetic inferences using RNA is complicated by alternative splicing, which produces multiple transcripts from a single gene. As such, a variety of approaches may be used to improve phylogenetic inference using transcriptomic data obtained from RNA-Seq and processed using computational phylogenetics.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.
Genome mining describes the exploitation of genomic information for the discovery of biosynthetic pathways of natural products and their possible interactions. It depends on computational technology and bioinformatics tools. The mining process relies on a huge amount of data accessible in genomic databases. By applying data mining algorithms, the data can be used to generate new knowledge in several areas of medicinal chemistry, such as discovering novel natural products.
References
- 1 2 Huerta-Cepas, Jaime; Capella-Gutierrez, S; Pryszcz, LP; Marcet-Houben, M; Gabaldón, T (Jan 2014). "PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome". Nucleic Acids Res. England. 42 (Database issue): D897–902. doi:10.1093/nar/gkt1177. PMC 3964985 . PMID 24275491.
- ↑ Huerta-Cepas, J; Bueno, A; Dopazo, J; Gabaldón, T (Jan 2008). "PhylomeDB: a database for genome-wide collections of gene phylogenies". Nucleic Acids Res. England. 36 (Database issue): D491–6. doi:10.1093/nar/gkm899. PMC 2238872 . PMID 17962297.
- ↑ Huerta-Cepas, Jaime; Capella-Gutierrez, S; Pryszcz, LP; Denisov, I; Kormes, D; Marcet-Houben, M; Gabaldón, T (Jan 2011). "PhylomeDB v3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions". Nucleic Acids Res. England. 39 (Database issue): D556–60. doi:10.1093/nar/gkq1109. PMC 3013701 . PMID 21075798.
- ↑ Capella-Gutierrez, S; Silla-Martínez, JM; Gabaldón, T (Aug 2009). "trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses". Bioinformatics. 25 (Database issue): 1972–3. doi:10.1093/bioinformatics/btp348. PMC 2712344 . PMID 19505945.
- ↑ Huerta-Cepas, J; Dopazo, J; Gabaldón, T (Jan 2010). "ETE: a python Environment for Tree Exploration". BMC Bioinformatics. 11 (Database issue): 24. doi:10.1186/1471-2105-11-24. PMC 2820433 . PMID 20070885.