
Carcinoma is a malignancy that develops from epithelial cells. Specifically, a carcinoma is a cancer that begins in a tissue that lines the inner or outer surfaces of the body, and that arises from cells originating in the endodermal, mesodermal or ectodermal germ layer during embryogenesis.
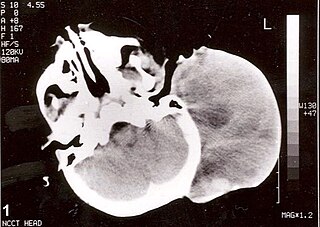
Rhabdomyosarcoma (RMS) is a highly aggressive form of cancer that develops from mesenchymal cells that have failed to fully differentiate into myocytes of skeletal muscle. Cells of the tumor are identified as rhabdomyoblasts.
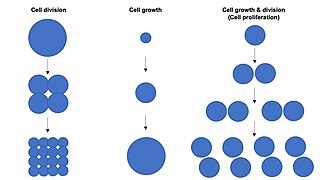
Cell growth refers to an increase in the total mass of a cell, including both cytoplasmic, nuclear and organelle volume. Cell growth occurs when the overall rate of cellular biosynthesis is greater than the overall rate of cellular degradation.

Invasive carcinoma of no special type, invasive breast carcinoma of no special type (IBC-NST), invasive ductal carcinoma (IDC), infiltrating ductal carcinoma (IDC) or invasive ductal carcinoma, not otherwise specified (NOS) is a disease. For international audiences this article will use "invasive carcinoma NST" because it is the preferred term of the World Health Organization (WHO).

Neuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine (hormonal) and nervous systems. They most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung, and the rest of the body.
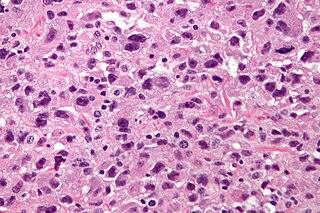
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.

Acinic cell carcinoma is a malignant tumor representing 2% of all salivary tumors. 90% of the time found in the parotid gland, 10% intraorally on buccal mucosa or palate. The disease presents as a slow growing mass, associated with pain or tenderness in 50% of the cases. Often appears pseudoencapsulated.
A mixed tumor is a tumor that derives from multiple tissue types. A biplastic tumor or biphasic tumor has two tissue types.

Fetal adenocarcinoma (FA) of the lung is a rare subtype of pulmonary adenocarcinoma that exhibits tissue architecture and cell characteristics that resemble fetal lung tissue upon microscopic examination. It is currently considered a variant of solid adenocarcinoma with mucin production.
Large cell lung carcinoma with rhabdoid phenotype (LCLC-RP) is a rare histological form of lung cancer, currently classified as a variant of large cell lung carcinoma (LCLC). In order for a LCLC to be subclassified as the rhabdoid phenotype variant, at least 10% of the malignant tumor cells must contain distinctive structures composed of tangled intermediate filaments that displace the cell nucleus outward toward the cell membrane. The whorled eosinophilic inclusions in LCLC-RP cells give it a microscopic resemblance to malignant cells found in rhabdomyosarcoma (RMS), a rare neoplasm arising from transformed skeletal muscle. Despite their microscopic similarities, LCLC-RP is not associated with rhabdomyosarcoma.
Mucinous cystadenocarcinoma of the lung (MCACL) is a very rare malignant mucus-producing neoplasm arising from the uncontrolled growth of transformed epithelial cells originating in lung tissue.
Embryonal rhabdomyosarcoma (EMRS) is a rare histological form of cancer in the connective tissue wherein the mesenchymally-derived malignant cells resemble the primitive developing skeletal muscle of the embryo. It is the most common soft tissue sarcoma occurring in children. Embryonal rhabdomyosarcoma is also known as PAX-fusion negative or fusion-negative rhabdomyosarcoma, as tumors of this subtype are unified by their lack of a PAX3-FOXO1 fusion oncogene. Fusion status refers to the presence or absence of a fusion gene, which is a gene formed from joining two different genes together through DNA rearrangements. These types of tumors are classified as embryonal rhabdomyosarcoma "because of their remarkable resemblance to developing embryonic and fetal skeletal muscle."
Sarcomatoid carcinoma of the lung is a term that encompasses five distinct histological subtypes of lung cancer, including (1) pleomorphic carcinoma, (2) spindle cell carcinoma, (3) giant cell carcinoma, (4) carcinosarcoma, or (5) pulmonary blastoma.

Giant-cell carcinoma of the lung (GCCL) is a rare histological form of large-cell lung carcinoma, a subtype of undifferentiated lung cancer, traditionally classified within the non-small-cell lung carcinomas (NSCLC).
Adenosquamous lung carcinoma (AdSqLC) is a biphasic malignant tumor arising from lung tissue that is composed of at least 10% by volume each of squamous cell carcinoma (SqCC) and adenocarcinoma (AdC) cells.
Salivary gland–like carcinomas of the lung generally refers a class of rare cancers that arise from the uncontrolled cell division (mitosis) of mutated cancer stem cells in lung tissue. They take their name partly from the appearance of their abnormal cells, whose structure and features closely resemble those of cancers that form in the major salivary glands of the head and neck. Carcinoma is a term for malignant neoplasms derived from cells of epithelial lineage, and/or that exhibit cytological or tissue architectural features characteristically found in epithelial cells.
Basaloid squamous cell carcinoma (Bas-SqCC) is an uncommon histological variant of lung cancer composed of cells exhibiting cytological and tissue architectural features of both squamous cell lung carcinoma and basal cell carcinoma.
Basaloid large cell carcinoma of the lung, is a rare histological variant of lung cancer featuring certain distinctive cytological, tissue architectural, and immunohistochemical characteristics and clinical behavior.

NUT carcinoma is a rare genetically defined, very aggressive squamous cell epithelial cancer that usually arises in the midline of the body and is characterized by a chromosomal rearrangement in the nuclear protein in testis gene. In approximately 75% of cases, the coding sequence of NUTM1 in band 14 on the long arm of chromosome 15 is fused to BRD4 or BRD3, which creates a chimeric gene that encodes the BRD-NUT fusion protein. The remaining cases, the fusion of NUTM1 is to an unknown partner gene, usually called NUT-variant.
A rhabdomyoblast is a cell type which is found in some rhabdomyosarcomas. When found histologically, a rhabdomyoblast aids the diagnosis of embryonal, alveolar, spindle cell/sclerosing, and pleomorphic rhabdomyosarcomas; however, in a tumor, expression of the rhabdomyoblast phenotype is not the only factor in diagnosing a rhabdomyosarcoma. Mesenchymal malignancies can exhibit this phenotype as well. Immunohistochemistry techniques allow for the sensitive detection of desmin, vimentin, muscle specific actin, and MyoD1. Similarly the rhabdomyoblast phenotype can be detected morphologically. Rhabdomyoblasts are early stage mesenchymal cells, having the potential to differentiate into a wide range of skeletal cells. Each stage of differentiation exhibits unique and distinguishable histological characteristics. In its initial from, stellate cells with amphiphilic cytoplasm and ovular central nuclei are observed. Commonly referred to as rhabdoid features, the maturing rhabdomyoblast will likely exhibit low levels of eosinophilic cytoplasm in proximal distances to the nucleus. As maturation and differentiation progress, the cell's cytoplasmic levels of white blood cells increase; additionally, elongated shapes, commonly depicted as “tadpole”, “strap” and "spider cells", are observed. In the concluding phase of differentiation, the white blood cell rich cytoplasm appears bright and exhibits cross-striation. The highly regulated organization of actin and myosin microfilaments in contractile proteins results in this appearance.
