Related Research Articles
Nuclear magnetic resonance spectroscopy of proteins is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins, and also nucleic acids, and their complexes. The field was pioneered by Richard R. Ernst and Kurt Wüthrich at the ETH, and by Ad Bax, Marius Clore, Angela Gronenborn at the NIH, and Gerhard Wagner at Harvard University, among others. Structure determination by NMR spectroscopy usually consists of several phases, each using a separate set of highly specialized techniques. The sample is prepared, measurements are made, interpretive approaches are applied, and a structure is calculated and validated.
The heteronuclear single quantum coherence or heteronuclear single quantum correlation experiment, normally abbreviated as HSQC, is used frequently in NMR spectroscopy of organic molecules and is of particular significance in the field of protein NMR. The experiment was first described by Geoffrey Bodenhausen and D. J. Ruben in 1980. The resulting spectrum is two-dimensional (2D) with one axis for proton (1H) and the other for a heteronucleus, which is usually 13C or 15N. The spectrum contains a peak for each unique proton attached to the heteronucleus being considered. The 2D HSQC can also be combined with other experiments in higher-dimensional NMR experiments, such as NOESY-HSQC or TOCSY-HSQC.

The Journal of Biomolecular NMR publishes research on technical developments and innovative applications of nuclear magnetic resonance spectroscopy for the study of structure and dynamic properties of biopolymers in solution, liquid crystals, solids and mixed environments. Some of the main topics include experimental and computational approaches for the determination of three-dimensional structures of proteins and nucleic acids, advancements in the automated analysis of NMR spectra, and new methods to probe and interpret molecular motions.
In biomolecular structure, CING stands for the Common Interface for NMR structure Generation and is known for structure and NMR data validation.
The Re-referenced Protein Chemical shift Database (RefDB) is an NMR spectroscopy database of carefully corrected or re-referenced chemical shifts, derived from the BioMagResBank (BMRB). The database was assembled by using a structure-based chemical shift calculation program to calculate expected protein (1)H, (13)C and (15)N chemical shifts from X-ray or NMR coordinate data of previously assigned proteins reported in the BMRB. The comparison is automatically performed by a program called SHIFTCOR. The RefDB database currently provides reference-corrected chemical shift data on more than 2000 assigned peptides and proteins. Data from the database indicates that nearly 25% of BMRB entries with (13)C protein assignments and 27% of BMRB entries with (15)N protein assignments require significant chemical shift reference readjustments. Additionally, nearly 40% of protein entries deposited in the BioMagResBank appear to have at least one assignment error. Users may download, search or browse the database through a number of methods available through the RefDB website. RefDB provides a standard chemical shift resource for biomolecular NMR spectroscopists, wishing to derive or compute chemical shift trends in peptides and proteins.
SHIFTCOR is a freely available web server as well as a stand-alone computer program for protein chemical shift re-referencing. Chemical shift referencing is a particularly widespread problem in biomolecular NMR with up to 25% of existing NMR chemical shift assignments being improperly referenced. Some of these referencing problems can lead to systematic errors of between 1.0 to 2.5 ppm. Errors of this magnitude can play havoc with any attempt to compare assignments between proteins or to structurally interpret chemical shifts. Identifying which proteins are mis-assigned or improperly referenced can be challenging, as can correcting the errors once they are found. The SHIFTCOR program was designed to assist with identifying and fixing these chemical shift referencing problems. Specifically it compares, identifies, corrects and re-references 1H, 13C and 15N backbone chemical shifts of peptides and proteins by comparing the observed chemical shifts with the predicted chemical shifts derived from the 3D structure of the protein(s) of interest [1]. The predicted chemical shifts are calculated using the ShiftX program. The SHIFTCOR program was originally used to construct a database of properly re-referenced protein chemical shift assignments called RefDB. RefDB is a web-accessible database of more than 2000 correctly referenced protein chemical shift assignments. While originally available as a stand-alone program only, SHIFTCOR has since been released for general use as a web server.

Random coil index (RCI) predicts protein flexibility by calculating an inverse weighted average of backbone secondary chemical shifts and predicting values of model-free order parameters as well as per-residue RMSD of NMR and molecular dynamics ensembles from this parameter.
Triple resonance experiments are a set of multi-dimensional nuclear magnetic resonance spectroscopy (NMR) experiments that link three types of atomic nuclei, most typically consisting of 1H, 15N and 13C. These experiments are often used to assign specific resonance signals to specific atoms in an isotopically-enriched protein. The technique was first described in papers by Ad Bax, Mitsuhiko Ikura and Lewis Kay in 1990, and further experiments were then added to the suite of experiments. Many of these experiments have since become the standard set of experiments used for sequential assignment of NMR resonances in the determination of protein structure by NMR. They are now an integral part of solution NMR study of proteins, and they may also be used in solid-state NMR.

GeNMR method is the first fully automated template-based method of protein structure determination that utilizes both NMR chemical shifts and NOE -based distance restraints.
Protein Structure Evaluation Suite & Server (PROSESS) is a freely available web server for protein structure validation. It has been designed at the University of Alberta to assist with the process of evaluating and validating protein structures solved by NMR spectroscopy.

CS23D is a web server to generate 3D structural models from NMR chemical shifts. CS23D combines maximal fragment assembly with chemical shift threading, de novo structure generation, chemical shift-based torsion angle prediction, and chemical shift refinement. CS23D makes use of RefDB and ShiftX.

The chemical shift index or CSI is a widely employed technique in protein nuclear magnetic resonance spectroscopy that can be used to display and identify the location as well as the type of protein secondary structure found in proteins using only backbone chemical shift data The technique was invented by David S. Wishart in 1992 for analyzing 1Hα chemical shifts and then later extended by him in 1994 to incorporate 13C backbone shifts. The original CSI method makes use of the fact that 1Hα chemical shifts of amino acid residues in helices tends to be shifted upfield relative to their random coil values and downfield in beta strands. Similar kinds of upfield and downfield trends are also detectable in backbone 13C chemical shifts.
Protein chemical shift prediction is a branch of biomolecular nuclear magnetic resonance spectroscopy that aims to accurately calculate protein chemical shifts from protein coordinates. Protein chemical shift prediction was first attempted in the late 1960s using semi-empirical methods applied to protein structures solved by X-ray crystallography. Since that time protein chemical shift prediction has evolved to employ much more sophisticated approaches including quantum mechanics, machine learning and empirically derived chemical shift hypersurfaces. The most recently developed methods exhibit remarkable precision and accuracy.
Nuclear magnetic resonance chemical shift re-referencing is a chemical analysis method for chemical shift referencing in biomolecular nuclear magnetic resonance (NMR). It has been estimated that up to 20% of 13C and up to 35% of 15N shift assignments are improperly referenced. Given that the structural and dynamic information contained within chemical shifts is often quite subtle, it is critical that protein chemical shifts be properly referenced so that these subtle differences can be detected. Fundamentally, the problem with chemical shift referencing comes from the fact that chemical shifts are relative frequency measurements rather than absolute frequency measurements. Because of the historic problems with chemical shift referencing, chemical shifts are perhaps the most precisely measurable but the least accurately measured parameters in all of NMR spectroscopy.
Protein chemical shift re-referencing is a post-assignment process of adjusting the assigned NMR chemical shifts to match IUPAC and BMRB recommended standards in protein chemical shift referencing. In NMR chemical shifts are normally referenced to an internal standard that is dissolved in the NMR sample. These internal standards include tetramethylsilane (TMS), 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) and trimethylsilyl propionate (TSP). For protein NMR spectroscopy the recommended standard is DSS, which is insensitive to pH variations. Furthermore, the DSS 1H signal may be used to indirectly reference 13C and 15N shifts using a simple ratio calculation [1]. Unfortunately, many biomolecular NMR spectroscopy labs use non-standard methods for determining the 1H, 13C or 15N “zero-point” chemical shift position. This lack of standardization makes it difficult to compare chemical shifts for the same protein between different laboratories. It also makes it difficult to use chemical shifts to properly identify or assign secondary structures or to improve their 3D structures via chemical shift refinement. Chemical shift re-referencing offers a means to correct these referencing errors and to standardize the reporting of protein chemical shifts across laboratories.
Resolution by Proxy (ResProx) is a method for assessing the equivalent X-ray resolution of NMR-derived protein structures. ResProx calculates resolution from coordinate data rather than from electron density or other experimental inputs. This makes it possible to calculate the resolution of a structure regardless of how it was solved. ResProx was originally designed to serve as a simple, single-number evaluation that allows straightforward comparison between the quality/resolution of X-ray structures and the quality of a given NMR structure. However, it can also be used to assess the reliability of an experimentally reported X-ray structure resolution, to evaluate protein structures solved by unconventional or hybrid means and to identify fraudulent structures deposited in the PDB. ResProx incorporates more than 25 different structural features to determine a single resolution-like value. ResProx values are reported in Angstroms. Tests on thousands of X-ray structures show that ResProx values match very closely to resolution values reported by X-ray crystallographers. Resolution-by-proxy values can be calculated for newly determined protein structures using a freely accessible ResProx web server. This server accepts protein coordinate data and generates a resolution estimate for that input structure.
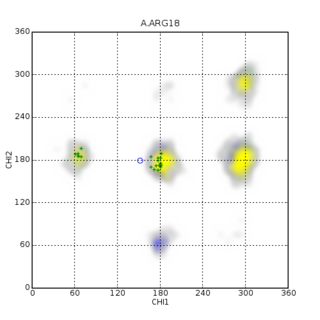
PREDITOR is a freely available web-server for the prediction of protein torsion angles from chemical shifts. For many years it has been known that protein chemical shifts are sensitive to protein secondary structure, which in turn, is sensitive to backbone torsion angles. torsion angles are internal coordinates that can be used to describe the conformation of a polypeptide chain. They can also be used as constraints to help determine or refine protein structures via NMR spectroscopy. In proteins there are four major torsion angles of interest: phi, psi, omega and chi-1. Traditionally protein NMR spectroscopists have used vicinal J-coupling information and the Karplus relation to determine approximate backbone torsion angle constraints for phi and chi-1 angles. However, several studies in the early 1990s pointed out the strong relationship between 1H and 13C chemical shifts and torsion angles, especially with backbone phi and psi angles. Later a number of other papers pointed out additional chemical shift relationships with chi-1 and even omega angles. PREDITOR was designed to exploit these experimental observations and to help NMR spectroscopists easily predict protein torsion angles from chemical shift assignments. Specifically, PREDITOR accepts protein sequence and/or chemical shift data as input and generates torsion angle predictions for phi, psi, omega and chi-1 angles. The algorithm that PREDITOR uses combines sequence alignment, chemical shift alignment and a number of related chemical shift analysis techniques to predict torsion angles. PREDITOR is unusually fast and exhibits a very high level of accuracy. In a series of tests 88% of PREDITOR’s phi/psi predictions were within 30 degrees of the correct values, 84% of chi-1 predictions were correct and 99.97% of PREDITOR’s predicted omega angles were correct. PREDITOR also estimates the torsion angle errors so that its torsion angle constraints can be used with standard protein structure refinement software, such as CYANA, CNS, XPLOR and AMBER. PREDITOR also supports automated protein chemical shift re-referencing and the prediction of proline cis/trans states. PREDITOR is not the only torsion angle prediction software available. Several other computer programs including TALOS, TALOS+ and DANGLE have also been developed to predict backbone torsion angles from protein chemical shifts. These stand-alone programs exhibit similar prediction performance to PREDITOR but are substantially slower.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of over 15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality, and identifying residue "outliers". The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR's output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.
ShiftX is a freely available web server for rapidly calculating protein chemical shifts from protein X-ray coordinates. Protein chemical shift prediction is particularly useful in verifying protein chemical shift assignments, adjusting mis-referenced chemical shifts, refining NMR protein structures and assisting with the NMR assignment of unassigned proteins that have either had their structures determined by X-ray or NMR methods.
David S. Wishart is a Canadian researcher in metabolomics and a Distinguished University Professor in the Department of Biological Sciences and the Department of Computing Science at the University of Alberta. Wishart also holds cross appointments in the Faculty of Pharmacy and Pharmaceutical Sciences and the Department of Laboratory Medicine and Pathology in the Faculty of Medicine and Dentistry. Additionally, Wishart holds a joint appointment in metabolomics at the Pacific Northwest National Laboratory in Richland, Washington. Wishart is well known for his pioneering contributions to the fields of protein NMR spectroscopy, bioinformatics, cheminformatics and metabolomics. In 2011, Wishart founded the Metabolomics Innovation Centre (TMIC), which is Canada's national metabolomics laboratory.
References
- 1 2 3 4 Wang, B; Wang, Y.; Wishart, D.S. (2010). "A probabilistic approach for validating protein NMR chemical shift assignments". J. Biomol. NMR. 47 (2): 85–99. doi:10.1007/s10858-010-9407-y. PMID 20446018. S2CID 22564072.
- 1 2 Zhang H, Neal S, Wishart DS (2003). "RefDB: A database of uniformly referenced protein chemical shifts". J. Biomol. NMR. 25 (3): 173–195. doi:10.1023/A:1022836027055. PMID 12652131. S2CID 12786364.
- ↑ Wushart, D.S. (Feb 2011). "Interpreting protein chemical shift data". Prog. Nucl. Magn. Reson. Spectrosc. 58 (1–2): 62–87. doi:10.1016/j.pnmrs.2010.07.004. PMID 21241884.