Related Research Articles

Gene therapy is a medical technology that aims to produce a therapeutic effect through the manipulation of gene expression or through altering the biological properties of living cells.

Leukemia is a group of blood cancers that usually begin in the bone marrow and result in high numbers of abnormal blood cells. These blood cells are not fully developed and are called blasts or leukemia cells. Symptoms may include bleeding and bruising, bone pain, fatigue, fever, and an increased risk of infections. These symptoms occur due to a lack of normal blood cells. Diagnosis is typically made by blood tests or bone marrow biopsy.
Canavan disease, or Canavan–Van Bogaert–Bertrand disease, is a rare and fatal autosomal recessive degenerative disease that causes progressive damage to nerve cells and loss of white matter in the brain. It is one of the most common degenerative cerebral diseases of infancy. It is caused by a deficiency of the enzyme aminoacylase 2, and is one of a group of genetic diseases referred to as leukodystrophies. It is characterized by degeneration of myelin in the phospholipid layer insulating the axon of a neuron and is associated with a gene located on human chromosome 17.
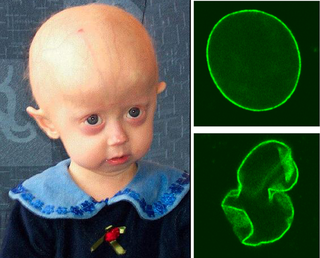
Progeria is a specific type of progeroid syndrome, also known as Hutchinson–Gilford syndrome. A single gene mutation is responsible for progeria. The gene, known as lamin A (LMNA), makes a protein necessary for holding the nucleus of the cell together. When this gene gets mutated, an abnormal form of lamin A protein called progerin is produced. Progeroid syndromes are a group of diseases that causes individuals to age faster than usual, leading to them appearing older than they actually are. Patients born with progeria typically live to an age of mid-teens to early twenties.
The National Eye Institute (NEI) is part of the U.S. National Institutes of Health (NIH), an agency of the U.S. Department of Health and Human Services. The mission of NEI is "to eliminate vision loss and improve quality of life through vision research." NEI consists of two major branches for research: an extramural branch that funds studies outside NIH and an intramural branch that funds research on the NIH campus in Bethesda, Maryland. Most of the NEI budget funds extramural research.

Chelation therapy is a medical procedure that involves the administration of chelating agents to remove heavy metals from the body. Chelation therapy has a long history of use in clinical toxicology and remains in use for some very specific medical treatments, although it is administered under very careful medical supervision due to various inherent risks, including the mobilization of mercury and other metals through the brain and other parts of the body by the use of weak chelating agents that unbind with metals before elimination, exacerbating existing damage. To avoid mobilization, some practitioners of chelation use strong chelators, such as selenium, taken at low doses over a long period of time.

Sanfilippo syndrome, also known as mucopolysaccharidosis type III (MPS III), is a rare autosomal recessive lysosomal storage disease that primarily affects the brain and spinal cord. It is caused by a buildup of large sugar molecules called glycosaminoglycans (AKA GAGs, or mucopolysaccharides) in the body's lysosomes.
Cognitive disorders (CDs), also known as neurocognitive disorders (NCDs), are a category of mental health disorders that primarily affect cognitive abilities including learning, memory, perception, and problem-solving. Neurocognitive disorders include delirium, mild neurocognitive disorders, and major neurocognitive disorder. They are defined by deficits in cognitive ability that are acquired, typically represent decline, and may have an underlying brain pathology. The DSM-5 defines six key domains of cognitive function: executive function, learning and memory, perceptual-motor function, language, complex attention, and social cognition.
The era of cancer chemotherapy began in the 1940s with the first use of nitrogen mustards and folic acid antagonist drugs. The targeted therapy revolution has arrived, but many of the principles and limitations of chemotherapy discovered by the early researchers still apply.
Metachromatic leukodystrophy (MLD) is a lysosomal storage disease which is commonly listed in the family of leukodystrophies as well as among the sphingolipidoses as it affects the metabolism of sphingolipids. Leukodystrophies affect the growth and/or development of myelin, the fatty covering which acts as an insulator around nerve fibers throughout the central and peripheral nervous systems. MLD involves cerebroside sulfate accumulation. Metachromatic leukodystrophy, like most enzyme deficiencies, has an autosomal recessive inheritance pattern.

Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.

X-linked agammaglobulinemia (XLA) is a rare genetic disorder discovered in 1952 that affects the body's ability to fight infection. As the form of agammaglobulinemia that is X-linked, it is much more common in males. In people with XLA, the white blood cell formation process does not generate mature B cells, which manifests as a complete or near-complete lack of proteins called gamma globulins, including antibodies, in their bloodstream. B cells are part of the immune system and normally manufacture antibodies, which defend the body from infections by sustaining a humoral immunity response. Patients with untreated XLA are prone to develop serious and even fatal infections. A mutation occurs at the Bruton's tyrosine kinase (Btk) gene that leads to a severe block in B cell development and a reduced immunoglobulin production in the serum. Btk is particularly responsible for mediating B cell development and maturation through a signaling effect on the B cell receptor BCR. Patients typically present in early childhood with recurrent infections, in particular with extracellular, encapsulated bacteria. XLA is deemed to have a relatively low incidence of disease, with an occurrence rate of approximately 1 in 200,000 live births and a frequency of about 1 in 100,000 male newborns. It has no ethnic predisposition. XLA is treated by infusion of human antibody. Treatment with pooled gamma globulin cannot restore a functional population of B cells, but it is sufficient to reduce the severity and number of infections due to the passive immunity granted by the exogenous antibodies.
Antonei Benjamin Csoka is a biogerontologist at Howard University who works on the molecular biology of aging, regenerative medicine, and epigenetics.

Elizabeth Nabel is an American cardiologist and Executive Vice President of Strategy at ModeX Therapeutics and OPKO Health. Prior to this role, she served as President of Brigham Health and its Brigham and Women's Hospital, Professor of Medicine at Harvard Medical School, and Director of the NIH's National Heart, Lung, and Blood Institute.

Laminopathies are a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina. They are included in the more generic term nuclear envelopathies that was coined in 2000 for diseases associated with defects of the nuclear envelope. Since the first reports of laminopathies in the late 1990s, increased research efforts have started to uncover the vital role of nuclear envelope proteins in cell and tissue integrity in animals.

Lonafarnib, sold under the brand name Zokinvy, is a medication used to reduce the risk of death due to Hutchinson-Gilford progeria syndrome and for the treatment of certain processing-deficient progeroid laminopathies in people one year of age and older.

Progerin is a truncated version of the lamin A protein involved in the pathology of Hutchinson–Gilford progeria syndrome. Progerin is most often generated by a sporadic single point nucleotide polymorphism c.1824 C>T in the gene that codes for matured Lamin A. This mutation activates a cryptic splice site that induces a mutation in premature Lamin A with the deletion of a 50 amino acids group near the C-terminus. The endopeptidase ZMPSTE24 cannot cleave between the missing RSY - LLG amino acid sequence during the maturation of Lamin A, due to the deletion of the 50 amino acids which included that sequence. This leaves the intact premature Lamin A bonded to the methylated carboxyl farnesyl group creating the defective protein Progerin, rather than the desired protein matured Lamin A. Approximately 90% of all Hutchinson–Gilford progeria syndrome cases are heterozygous for this deleterious single nucleotide polymorphism within exon 11 of the LMNA gene causing the post-translational modifications to produce Progerin.

Hayley Leanne Okines was an English author and activist who was a sufferer of the extremely rare aging disease progeria. She was known for spreading awareness of the condition. Although the average life expectancy for those with the condition is 13 years, Okines was part of a drug trial that had seen her surpass doctors' predictions of her projected lifespan. She died on 2 April 2015 at the age of 17, having lived four years beyond doctors' initial predictions.

Sampson GordonBerns was an American activist with progeria, an extremely rare and fatal disease that causes the body to age rapidly. Berns helped raise awareness about the disease and he was the subject of the HBO documentary Life According to Sam, which was first screened in January 2013.

Life According to Sam is an HBO original documentary film directed by Sean Fine and Andrea Nix Fine. Premiering in January 2013 at the Sundance Film Festival, the documentary discloses the impact that progeria had on the lives of Sam Berns and his parents, Dr. Leslie Gordon and Dr. Scott Berns. It was broadcast on HBO in October 2013, and since then it has won a 2013 Peabody Award and an Emmy Award for Exceptional Merit in Documentary Filmmaking. It was also one of the 15 titles considered for nomination in the Documentary Feature category for the 86th Oscars.
References
- 1 2 Moore, Keith. Old at Age 3: The Story of Zachary Moore Old At Age 3, 2007 ISBN 061516062X, pp. 11–13
- ↑ Klatz, Ronald. The New Anti-aging Revolution: Stopping the Clock for a Younger, Sexier, Happier You! ReadHowYouWant.com, 2009. ISBN 1458716228, p. 274
- ↑ "FDA Approves Drug for Progeria, a Rare Disease Causing Rapid Aging in Children". AJMC. Retrieved 2021-01-17.
- ↑ Gordon, Leslie B.; Shappell, Heather; Massaro, Joe; D’Agostino, Ralph B.; Brazier, Joan; Campbell, Susan E.; Kleinman, Monica E.; Kieran, Mark W. (2018-04-24). "Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome". JAMA. 319 (16): 1687. doi: 10.1001/jama.2018.3264 . ISSN 0098-7484.