
Nuclear magnetic resonance spectroscopy, most commonly known as NMR spectroscopy or magnetic resonance spectroscopy (MRS), is a spectroscopic technique based on re-orientation of atomic nuclei with non-zero nuclear spins in an external magnetic field. This re-orientation occurs with absorption of electromagnetic radiation in the radio frequency region from roughly 4 to 900 MHz, which depends on the isotopic nature of the nucleus and increased proportionally to the strength of the external magnetic field. Notably, the resonance frequency of each NMR-active nucleus depends on its chemical environment. As a result, NMR spectra provide information about individual functional groups present in the sample, as well as about connections between nearby nuclei in the same molecule. As the NMR spectra are unique or highly characteristic to individual compounds and functional groups, NMR spectroscopy is one of the most important methods to identify molecular structures, particularly of organic compounds.
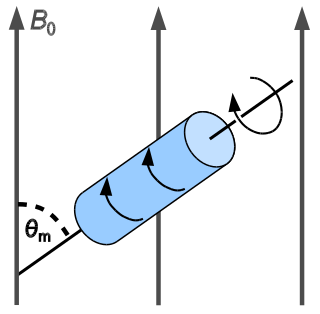
In solid-state NMR spectroscopy, magic-angle spinning (MAS) is a technique routinely used to produce better resolution NMR spectra. MAS NMR consists in spinning the sample at the magic angle θm with respect to the direction of the magnetic field.

Solid-state nuclear magnetic resonance (ssNMR) is a spectroscopy technique used to characterize atomic-level structure and dynamics in solid materials. ssNMR spectra are broader due to nuclear spin interactions which can be categorized as dipolar coupling, chemical shielding, quadrupolar interactions, and j-coupling. These interactions directly affect the lines shapes of experimental ssNMR spectra which can be seen in powder and dipolar patterns. There are many essential solid-state techniques alongside advanced ssNMR techniques that may be applied to elucidate the fundamental aspects of solid materials. ssNMR is often combined with magic angle spinning (MAS) to remove anisotropic interactions and improve the sensitivity of the technique. The applications of ssNMR further extend to biology and medicine.
Nuclear magnetic resonance spectroscopy of proteins is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins, and also nucleic acids, and their complexes. The field was pioneered by Richard R. Ernst and Kurt Wüthrich at the ETH, and by Ad Bax, Marius Clore, Angela Gronenborn at the NIH, and Gerhard Wagner at Harvard University, among others. Structure determination by NMR spectroscopy usually consists of several phases, each using a separate set of highly specialized techniques. The sample is prepared, measurements are made, interpretive approaches are applied, and a structure is calculated and validated.
Xplor-NIH is a highly sophisticated and flexible biomolecular structure determination program which includes an interface to the legacy X-PLOR program. The main developers are Charles Schwieters and Marius Clore of the National Institutes of Health. Xplor-NIH is based on a C++ framework with an extensive Python interface enabling very powerful and easy scripting of complex structure determination and refinement protocols. Restraints derived from all current solution and many solid state nuclear magnetic resonance (NMR) and X-ray scattering experiments can be accommodated during structure calculations. Extensive facilities are also available for many types of ensemble calculations where the experimental data cannot be accounted for by a unique structure. Many of the structure calculation protocols involve the use of simulated annealing designed to overcome local minima on the path of the global minimum region of the target function. These calculations can be carried out using any combination of Cartesian, torsion angle and rigid body dynamics and minimization. Currently Xplor-NIH is the most versatile, comprehensive and widely used structure determination/refinement package in NMR structure determination.
Transverse relaxation optimized spectroscopy (TROSY) is an experiment in protein NMR spectroscopy that allows studies of large molecules or complexes.
In nuclear chemistry and nuclear physics, J-couplings are mediated through chemical bonds connecting two spins. It is an indirect interaction between two nuclear spins that arises from hyperfine interactions between the nuclei and local electrons. In NMR spectroscopy, J-coupling contains information about relative bond distances and angles. Most importantly, J-coupling provides information on the connectivity of chemical bonds. It is responsible for the often complex splitting of resonance lines in the NMR spectra of fairly simple molecules.

The residual dipolar coupling between two spins in a molecule occurs if the molecules in solution exhibit a partial alignment leading to an incomplete averaging of spatially anisotropic dipolar couplings.
Adriaan "Ad" Bax is a Dutch-American molecular biophysicist. He was born in the Netherlands and is the Chief of the Section on Biophysical NMR Spectroscopy at the National Institutes of Health. He is known for his work on the methodology of biomolecular NMR spectroscopy. He is a corresponding member of the Royal Netherlands Academy of Arts and Sciences, a member of the National Academy of Sciences, a fellow of the American Academy of Arts and Sciences, and a Foreign Member of the Royal Society.

Nuclear magnetic resonance (NMR) is a physical phenomenon in which nuclei in a strong constant magnetic field are disturbed by a weak oscillating magnetic field and respond by producing an electromagnetic signal with a frequency characteristic of the magnetic field at the nucleus. This process occurs near resonance, when the oscillation frequency matches the intrinsic frequency of the nuclei, which depends on the strength of the static magnetic field, the chemical environment, and the magnetic properties of the isotope involved; in practical applications with static magnetic fields up to ca. 20 tesla, the frequency is similar to VHF and UHF television broadcasts (60–1000 MHz). NMR results from specific magnetic properties of certain atomic nuclei. High-resolution nuclear magnetic resonance spectroscopy is widely used to determine the structure of organic molecules in solution and study molecular physics and crystals as well as non-crystalline materials. NMR is also routinely used in advanced medical imaging techniques, such as in magnetic resonance imaging (MRI). The original application of NMR to condensed matter physics is nowadays mostly devoted to strongly correlated electron systems. It reveals large many-body couplings by fast broadband detection and should not be confused with solid state NMR, which aims at removing the effect of the same couplings by Magic Angle Spinning techniques.
Nuclear magnetic resonance crystallography is a method which utilizes primarily NMR spectroscopy to determine the structure of solid materials on the atomic scale. Thus, solid-state NMR spectroscopy would be used primarily, possibly supplemented by quantum chemistry calculations, powder diffraction etc. If suitable crystals can be grown, any crystallographic method would generally be preferred to determine the crystal structure comprising in case of organic compounds the molecular structures and molecular packing. The main interest in NMR crystallography is in microcrystalline materials which are amenable to this method but not to X-ray, neutron and electron diffraction. This is largely because interactions of comparably short range are measured in NMR crystallography.
Gary Martin is an American chemist and expert in the fields of both NMR spectroscopy and medicinal chemistry. He is a distinguished fellow at the Merck Research Laboratories. He is also a photographer specializing in the capture of images of lighthouses, especially under conditions of extreme weather.
Nucleic acid NMR is the use of nuclear magnetic resonance spectroscopy to obtain information about the structure and dynamics of nucleic acid molecules, such as DNA or RNA. It is useful for molecules of up to 100 nucleotides, and as of 2003, nearly half of all known RNA structures had been determined by NMR spectroscopy.
CS-ROSETTA is a framework for structure calculation of biological macromolecules on the basis of conformational information from NMR, which is built on top of the biomolecular modeling and design software called ROSETTA. The name CS-ROSETTA for this branch of ROSETTA stems from its origin in combining NMR chemical shift (CS) data with ROSETTA structure prediction protocols. The software package was later extended to include additional NMR conformational parameters, such as Residual Dipolar Couplings (RDC), NOE distance restraints, pseudocontact chemical shifts (PCS) and restraints derived from homologous proteins. This software can be used together with other molecular modeling protocols, such as docking to model protein oligomers. In addition, CS-ROSETTA can be combined with chemical shift resonance assignment algorithms to create a fully automated NMR structure determination pipeline. The CS-ROSETTA software is freely available for academic use and can be licensed for commercial use. A software manual and tutorials are provided on the supporting website https://csrosetta.chemistry.ucsc.edu/.
Triple resonance experiments are a set of multi-dimensional nuclear magnetic resonance spectroscopy (NMR) experiments that link three types of atomic nuclei, most typically consisting of 1H, 15N and 13C. These experiments are often used to assign specific resonance signals to specific atoms in an isotopically-enriched protein. The technique was first described in papers by Ad Bax, Mitsuhiko Ikura and Lewis Kay in 1990, and further experiments were then added to the suite of experiments. Many of these experiments have since become the standard set of experiments used for sequential assignment of NMR resonances in the determination of protein structure by NMR. They are now an integral part of solution NMR study of proteins, and they may also be used in solid-state NMR.

Paramagnetic nuclear magnetic resonance spectroscopy refers to nuclear magnetic resonance (NMR) spectroscopy of paramagnetic compounds. Although most NMR measurements are conducted on diamagnetic compounds, paramagnetic samples are also amenable to analysis and give rise to special effects indicated by a wide chemical shift range and broadened signals. Paramagnetism diminishes the resolution of an NMR spectrum to the extent that coupling is rarely resolved. Nonetheless spectra of paramagnetic compounds provide insight into the bonding and structure of the sample. For example, the broadening of signals is compensated in part by the wide chemical shift range (often 200 ppm in 1H NMR). Since paramagnetism leads to shorter relaxation times (T1), the rate of spectral acquisition can be high.
Protein Structure Evaluation Suite & Server (PROSESS) is a freely available web server for protein structure validation. It has been designed at the University of Alberta to assist with the process of evaluating and validating protein structures solved by NMR spectroscopy.

In computational chemistry, conformational ensembles, also known as structural ensembles, are experimentally constrained computational models describing the structure of intrinsically unstructured proteins. Such proteins are flexible in nature, lacking a stable tertiary structure, and therefore cannot be described with a single structural representation. The techniques of ensemble calculation are relatively new on the field of structural biology, and are still facing certain limitations that need to be addressed before it will become comparable to classical structural description methods such as biological macromolecular crystallography.
Protein chemical shift prediction is a branch of biomolecular nuclear magnetic resonance spectroscopy that aims to accurately calculate protein chemical shifts from protein coordinates. Protein chemical shift prediction was first attempted in the late 1960s using semi-empirical methods applied to protein structures solved by X-ray crystallography. Since that time protein chemical shift prediction has evolved to employ much more sophisticated approaches including quantum mechanics, machine learning and empirically derived chemical shift hypersurfaces. The most recently developed methods exhibit remarkable precision and accuracy.

G. Marius Clore MAE, FRSC, FMedSci, FRS is a British-born, Anglo-American molecular biophysicist and structural biologist. He was born in London, U.K. and is a dual U.S./U.K. Citizen. He is a Member of the National Academy of Sciences, a Fellow of the Royal Society, a Fellow of the Academy of Medical Sciences, a Fellow of the American Academy of Arts and Sciences, a NIH Distinguished Investigator, and the Chief of the Molecular and Structural Biophysics Section in the Laboratory of Chemical Physics of the National Institute of Diabetes and Digestive and Kidney Diseases at the U.S. National Institutes of Health. He is known for his foundational work in three-dimensional protein and nucleic acid structure determination by biomolecular NMR spectroscopy, for advancing experimental approaches to the study of large macromolecules and their complexes by NMR, and for developing NMR-based methods to study rare conformational states in protein-nucleic acid and protein-protein recognition. Clore's discovery of previously undetectable, functionally significant, rare transient states of macromolecules has yielded fundamental new insights into the mechanisms of important biological processes, and in particular the significance of weak interactions and the mechanisms whereby the opposing constraints of speed and specificity are optimized. Further, Clore's work opens up a new era of pharmacology and drug design as it is now possible to target structures and conformations that have been heretofore unseen.