Related Research Articles

In biology, histones are highly basic proteins abundant in lysine and arginine residues that are found in eukaryotic cell nuclei. They act as spools around which DNA winds to create structural units called nucleosomes. Nucleosomes in turn are wrapped into 30-nanometer fibers that form tightly packed chromatin. Histones prevent DNA from becoming tangled and protect it from DNA damage. In addition, histones play important roles in gene regulation and DNA replication. Without histones, unwound DNA in chromosomes would be very long. For example, each human cell has about 1.8 meters of DNA if completely stretched out; however, when wound about histones, this length is reduced to about 90 micrometers (0.09 mm) of 30 nm diameter chromatin fibers.

Histone methyltransferases (HMT) are histone-modifying enzymes, that catalyze the transfer of one, two, or three methyl groups to lysine and arginine residues of histone proteins. The attachment of methyl groups occurs predominantly at specific lysine or arginine residues on histones H3 and H4. Two major types of histone methyltranferases exist, lysine-specific and arginine-specific. In both types of histone methyltransferases, S-Adenosyl methionine (SAM) serves as a cofactor and methyl donor group.
The genomic DNA of eukaryotes associates with histones to form chromatin. The level of chromatin compaction depends heavily on histone methylation and other post-translational modifications of histones. Histone methylation is a principal epigenetic modification of chromatin that determines gene expression, genomic stability, stem cell maturation, cell lineage development, genetic imprinting, DNA methylation, and cell mitosis.

RNA polymerase II is a multiprotein complex that transcribes DNA into precursors of messenger RNA (mRNA) and most small nuclear RNA (snRNA) and microRNA. It is one of the three RNAP enzymes found in the nucleus of eukaryotic cells. A 550 kDa complex of 12 subunits, RNAP II is the most studied type of RNA polymerase. A wide range of transcription factors are required for it to bind to upstream gene promoters and begin transcription.
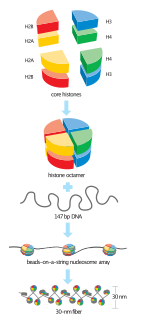
Histone H4 is one of the five main histone proteins involved in the structure of chromatin in eukaryotic cells. Featuring a main globular domain and a long N-terminal tail, H4 is involved with the structure of the nucleosome of the 'beads on a string' organization. Histone proteins are highly post-translationally modified. Covalently bonded modifications include acetylation and methylation of the N-terminal tails. These modifications may alter expression of genes located on DNA associated with its parent histone octamer. Histone H4 is an important protein in the structure and function of chromatin, where its sequence variants and variable modification states are thought to play a role in the dynamic and long term regulation of genes.
Histone methylation is a process by which methyl groups are transferred to amino acids of histone proteins that make up nucleosomes, which the DNA double helix wraps around to form chromosomes. Methylation of histones can either increase or decrease transcription of genes, depending on which amino acids in the histones are methylated, and how many methyl groups are attached. Methylation events that weaken chemical attractions between histone tails and DNA increase transcription because they enable the DNA to uncoil from nucleosomes so that transcription factor proteins and RNA polymerase can access the DNA. This process is critical for the regulation of gene expression that allows different cells to express different genes.

Methyltransferases are a large group of enzymes that all methylate their substrates but can be split into several subclasses based on their structural features. The most common class of methyltransferases is class I, all of which contain a Rossmann fold for binding S-Adenosyl methionine (SAM). Class II methyltransferases contain a SET domain, which are exemplified by SET domain histone methyltransferases, and class III methyltransferases, which are membrane associated. Methyltransferases can also be grouped as different types utilizing different substrates in methyl transfer reactions. These types include protein methyltransferases, DNA/RNA methyltransferases, natural product methyltransferases, and non-SAM dependent methyltransferases. SAM is the classical methyl donor for methyltransferases, however, examples of other methyl donors are seen in nature. The general mechanism for methyl transfer is a SN2-like nucleophilic attack where the methionine sulfur serves as the leaving group and the methyl group attached to it acts as the electrophile that transfers the methyl group to the enzyme substrate. SAM is converted to S-Adenosyl homocysteine (SAH) during this process. The breaking of the SAM-methyl bond and the formation of the substrate-methyl bond happen nearly simultaneously. These enzymatic reactions are found in many pathways and are implicated in genetic diseases, cancer, and metabolic diseases. Another type of methyl transfer is the radical S-Adenosyl methionine (SAM) which is the methylation of unactivated carbon atoms in primary metabolites, proteins, lipids, and RNA.

Histone-modifying enzymes are enzymes involved in the modification of histone substrates after protein translation and affect cellular processes including gene expression. To safely store the eukaryotic genome, DNA is wrapped around four core histone proteins, which then join to form nucleosomes. These nucleosomes further fold together into highly condensed chromatin, which renders the organism's genetic material far less accessible to the factors required for gene transcription, DNA replication, recombination and repair. Subsequently, eukaryotic organisms have developed intricate mechanisms to overcome this repressive barrier imposed by the chromatin through histone modification, a type of post-translational modification which typically involves covalently attaching certain groups to histone residues. Once added to the histone, these groups elicit either a loose and open histone conformation, euchromatin, or a tight and closed histone conformation, heterochromatin. Euchromatin marks active transcription and gene expression, as the light packing of histones in this way allows entry for proteins involved in the transcription process. As such, the tightly packed heterochromatin marks the absence of current gene expression.
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, e.g., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes. Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, egg cells DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.

Enhancer of zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase enzyme encoded by EZH2 gene, that participates in histone methylation and, ultimately, transcriptional repression. EZH2 catalyzes the addition of methyl groups to histone H3 at lysine 27, by using the cofactor S-adenosyl-L-methionine. Methylation activity of EZH2 facilitates heterochromatin formation thereby silences gene function. Remodeling of chromosomal heterochromatin by EZH2 is also required during cell mitosis.

Histone-lysine N-methyltransferase SUV39H1 is an enzyme that in humans is encoded by the SUV39H1 gene.

Histone-lysine N-methyltransferase 2A also known as acute lymphoblastic leukemia 1 (ALL-1), myeloid/lymphoid or mixed-lineage leukemia1 (MLL1), or zinc finger protein HRX (HRX) is an enzyme that in humans is encoded by the KMT2A gene.

DOT1-like, histone H3K79 methyltransferase, also known as DOT1L, is a protein found in humans, as well as other eukaryotes. The methylation of histone H3 lysine 79 (H3K79) by DOT1L which is a conserved epigenetic mark in many eukaryotic epigenomes, increases progressively along the aging process, suggesting that "DOT1L might function as a vital clock, ticking the hours impassively".
Cryptic unstable transcripts (CUTs) are a subset of non-coding RNAs (ncRNAs) that are produced from intergenic and intragenic regions. CUTs were first observed in S. cerevisiae yeast models and are found in most eukaryotes. Some basic characteristics of CUTs include a length of around 200–800 base pairs, a 5' cap, poly-adenylated tail, and rapid degradation due to the combined activity of poly-adenylating polymerases and exosome complexes. CUT transcription occurs through RNA Polymerase II and initiates from nucleosome-depleted regions, often in an antisense orientation. To date, CUTs have a relatively uncharacterized function but have been implicated in a number of putative gene regulation and silencing pathways. Thousands of loci leading to the generation of CUTs have been described in the yeast genome. Additionally, stable uncharacterized transcripts, or SUTs, have also been detected in cells and bear many similarities to CUTs but are not degraded through the same pathways.

The SET domain is a protein domain that typically has methyltransferase activity. It was originally identified as part of a larger conserved region present in the Drosophila Trithorax protein and was subsequently identified in the Drosophila Su(var)3-9 and 'Enhancer of zeste' proteins, from which the acronym SET is derived [Su(var)3-9, Enhancer-of-zeste and Trithorax].

Euchromatic histone-lysine N-methyltransferase 1, also known as G9a-like protein (GLP), is a protein that in humans is encoded by the EHMT1 gene.

Ali Shilatifard is an American biochemist/molecular biologist, the Robert Francis Furchgott Professor and Chairman of the department of biochemistry and molecular genetics, and the director of the Simpson Query Institute for Epigenetics at the Northwestern University Feinberg School of Medicine. He has served as a senior editor for the journal Science, and currently serves as the Editor for Science's open access journal Science Advances. Research in Shilatifard's lab focuses on the cause of childhood leukemia through chromosomal translocations, the role of ELL in this process, and the discovery of the Super Elongation Complex as being a central complex linking MLL translocations into a diverse number of genes to leukemic pathogenesis.
SilentInformationRegulator (SIR) proteins are involved in regulating gene expression. SIR proteins organize heterochromatin near telomeres, rDNA, and at silent loci including hidden mating type loci in yeast. The SIR family of genes encodes catalytic and non-catalytic proteins that are involved in de-acetylation of histone tails and the subsequent condensation of chromatin around a SIR protein scaffold. Some SIR family members are conserved from yeast to humans.
Protein methylation is a type of post-translational modification featuring the addition of methyl groups to proteins. It can occur on the nitrogen-containing side-chains of arginine and lysine, but also at the amino- and carboxy-termini of a number of different proteins. In biology, methyltransferases catalyze the methylation process, activated primarily by S-adenosylmethionine. Protein methylation has been most studied in histones, where the transfer of methyl groups from S-adenosyl methionine is catalyzed by histone methyltransferases. Histones that are methylated on certain residues can act epigenetically to repress or activate gene expression.

Thomas Jenuwein is a German scientist working in the fields of epigenetics, chromatin biology, gene regulation and genome function.
In epigenetics, proline isomerization is the effect that cis-trans isomerization of the amino acid proline has on the regulation of gene expression. Similar to aspartic acid, the amino acid proline has the rare property of being able to occupy both cis and trans isomers of its prolyl peptide bonds with ease. Peptidyl-prolyl isomerase, or PPIase, is an enzyme very commonly associated with proline isomerization due to their ability to catalyze the isomerization of prolines. PPIases are present in three types: cyclophilins, FK507-binding proteins, and the parvulins. PPIase enzymes catalyze the transition of proline between cis and trans isomers and are essential to the numerous biological functions controlled and affected by prolyl isomerization Without PPIases, prolyl peptide bonds will slowly switch between cis and trans isomers, a process that can lock proteins in a nonnative structure that can affect render the protein temporarily ineffective. Although this switch can occur on its own, PPIases are responsible for most isomerization of prolyl peptide bonds. The specific amino acid that precedes the prolyl peptide bond also can have an effect on which conformation the bond assumes. For instance, when an aromatic amino acid is bonded to a proline the bond is more favorable to the cis conformation. Cyclophilin A uses an "electrostatic handle" to pull proline into cis and trans formations. Most of these biological functions are affected by the isomerization of proline when one isomer interacts differently than the other, commonly causing an activation/deactivation relationship. As an amino acid, proline is present in many proteins. This aids in the multitude of effects that isomerization of proline can have in different biological mechanisms and functions.
References
- 1 2 3 "SET1 - Histone-lysine N-methyltransferase, H3 lysine-4 specific - Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) - SET1 gene & protein". www.uniprot.org. Retrieved 2022-04-26.
- ↑ Shilatifard, Ali (2012). "The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis". Annual review of biochemistry. 81: 65–95. doi:10.1146/annurev-biochem-051710-134100. ISSN 0066-4154. PMC 4010150 . PMID 22663077.
- ↑ Briggs, Scott D.; Bryk, Mary; Strahl, Brian D.; Cheung, Wang L.; Davie, Judith K.; Dent, Sharon Y. R.; Winston, Fred; Allis, C. David (2001-12-15). "Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae". Genes & Development. 15 (24): 3286–3295. doi:10.1101/gad.940201. ISSN 0890-9369. PMID 11751634.
| | This gene article is a stub. You can help Wikipedia by expanding it. |