A nucleosome is a basic unit of DNA packaging in eukaryotes, consisting of a segment of DNA wound in sequence around eight histone protein cores. This structure is often compared to thread wrapped around a spool.

An epigenome consists of a record of the chemical changes to the DNA and histone proteins of an organism; these changes can be passed down to an organism's offspring via transgenerational epigenetic inheritance. Changes to the epigenome can result in changes to the structure of chromatin and changes to the function of the genome.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
DamID is a molecular biology protocol used to map the binding sites of DNA- and chromatin-binding proteins in eukaryotes. DamID identifies binding sites by expressing the proposed DNA-binding protein as a fusion protein with DNA methyltransferase. Binding of the protein of interest to DNA localizes the methyltransferase in the region of the binding site. Adenosine methylation does not occur naturally in eukaryotes and therefore adenine methylation in any region can be concluded to have been caused by the fusion protein, implying the region is located near a binding site. DamID is an alternate method to ChIP-on-chip or ChIP-seq.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
Chromatin Interaction Analysis by Paired-End Tag Sequencing is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.
Genes can be regulated by regions far from the promoter such as regulatory elements, insulators and boundary elements, and transcription-factor binding sites (TFBS). Uncovering the interplay between regulatory regions and gene coding regions is essential for understanding the mechanisms governing gene regulation in health and disease. ChIA-PET can be used to identify unique, functional chromatin interactions between distal and proximal regulatory transcription-factor binding sites and the promoters of the genes they interact with.

Chromatin immunoprecipitation (ChIP) is a type of immunoprecipitation experimental technique used to investigate the interaction between proteins and DNA in the cell. It aims to determine whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites, and possibly defining cistromes. ChIP also aims to determine the specific location in the genome that various histone modifications are associated with, indicating the target of the histone modifiers.
FAIRE-Seq is a method in molecular biology used for determining the sequences of DNA regions in the genome associated with regulatory activity. The technique was developed in the laboratory of Jason D. Lieb at the University of North Carolina, Chapel Hill. In contrast to DNase-Seq, the FAIRE-Seq protocol doesn't require the permeabilization of cells or isolation of nuclei, and can analyse any cell type. In a study of seven diverse human cell types, DNase-seq and FAIRE-seq produced strong cross-validation, with each cell type having 1-2% of the human genome as open chromatin.
DNase-seq is a method in molecular biology used to identify the location of regulatory regions, based on the genome-wide sequencing of regions sensitive to cleavage by DNase I. FAIRE-Seq is a successor of DNase-seq for the genome-wide identification of accessible DNA regions in the genome. Both the protocols for identifying open chromatin regions have biases depending on underlying nucleosome structure. For example, FAIRE-seq provides higher tag counts at non-promoter regions. On the other hand, DNase-seq signal is higher at promoter regions, and DNase-seq has been shown to have better sensitivity than FAIRE-seq even at non-promoter regions.
ChiRP-Seq is a high-throughput sequencing method to discover regions of the genome which are bound by a specific RNA. Recent studies have shown that a significant proportion of some genomes synthesize RNA that apparently do not code for proteins. The function of most of these non-coding RNA still has to be ascertained. Various genomic methods are being developed to map the functional association of these novel RNA to distinct regions of the genome to gain a better understanding of their function. ChiRP-Seq is one of these new methods which uses the massively parallel sequencing capability of 2nd generation sequencers to catalog the binding sites of these novel RNA molecules on a genome.
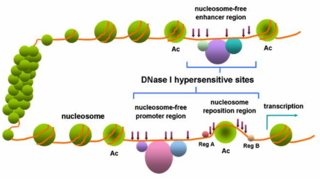
In genetics, DNase I hypersensitive sites (DHSs) are regions of chromatin that are sensitive to cleavage by the DNase I enzyme. In these specific regions of the genome, chromatin has lost its condensed structure, exposing the DNA and making it accessible. This raises the availability of DNA to degradation by enzymes, such as DNase I. These accessible chromatin zones are functionally related to transcriptional activity, since this remodeled state is necessary for the binding of proteins such as transcription factors.
Enhancer RNAs (eRNAs) represent a class of relatively short non-coding RNA molecules transcribed from the DNA sequence of enhancer regions. They were first detected in 2010 through the use of genome-wide techniques such as RNA-seq and ChIP-seq. eRNAs can be subdivided into two main classes: 1D eRNAs and 2D eRNAs, which differ primarily in terms of their size, polyadenylation state, and transcriptional directionality. The expression of a given eRNA seems to correlate with the activity of its corresponding enhancer in a context-dependent fashion. Increasing evidence suggests that eRNAs actively play a role in transcriptional regulation in cis and in trans, and while their mechanisms of action remain unclear, a few models have been proposed.
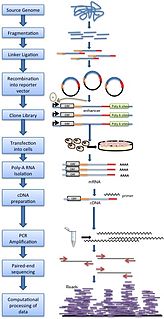
STARR-seq is a novel method to assay enhancer activity for millions of candidates from arbitrary sources of DNA. It is used to identify the sequences that act as transcriptional enhancers in a direct, quantitative, and genome-wide manner.
Chem-seq is a technique that is used to map genome-wide interactions between small molecules and their protein targets in the chromatin of eukaryotic cell nuclei. The method employs chemical affinity capture coupled with massively parallel DNA sequencing to identify genomic sites where small molecules interact with their target proteins or DNA. It was first described by Lars Anders et al. in the January, 2014 issue of "Nature Biotechnology".
ATAC-seq is a technique used in molecular biology to study chromatin accessibility. The technique was first described in 2013, as an alternative or complementary method to MNase-seq, FAIRE-seq and DNAse-seq. It aims to identify accessible DNA regions, equivalent to DNase I hypersensitive sites.
H3K4me3 is an epigenetic chemical modification involved in the regulation of gene expression. The name denotes the addition of three methyl groups (trimethylation) to the lysine 4 on the histone H3 protein. H3 is used to package DNA in eukaryotic cells, and modifications to the histone alter the accessibility of genes for transcription. H3K4me3 is commonly associated with the activation of transcription of nearby genes. H3K4 trimethylation regulates gene expression through chromatin remodeling by the NURF complex. In bivalent chromatin, H3K4me3 is co-localized with the repressive modification H3K27me3 to control gene regulation. H3K4me3 also plays an important role in the genetic regulation of stem cell potency and lineage.
H3K27me3 is a histone methylation occurring on the amino (N) terminal tail of the core histone H3. This tri-methylation is associated with the downregulation of nearby genes via the formation of heterochromatic regions.

Single cell epigenomics is the study of epigenomics in individual cells by single cell sequencing. Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.
CUT&RUN-sequencing, also known as cleavage under targets and release using nuclease, is a method used to analyze protein interactions with DNA. CUT&RUN-sequencing combines antibody-targeted controlled cleavage by micrococcal nuclease with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN-Sequencing does not.
