A sarcoma is a cancer that arises from transformed cells of mesenchymal origin. Connective tissue is a broad term that includes bone, cartilage, fat, vascular, or hematopoietic tissues, and sarcomas can arise in any of these types of tissues. As a result, there are many subtypes of sarcoma, which are classified based on the specific tissue and type of cell from which the tumor originates. It is important to note that sarcomas are primary connective tissue tumors, meaning that they arise in connective tissues. This is in contrast to secondary connective tissue tumors, which occur when a cancer from elsewhere in the body spreads to the connective tissue. The word sarcoma is derived from the Greek σάρξ sarx meaning "flesh".

Li–Fraumeni syndrome is a rare, autosomal dominant, hereditary disorder that predisposes carriers to cancer development. It was named after two American physicians, Frederick Pei Li and Joseph F. Fraumeni, Jr., who first recognized the syndrome after reviewing the medical records and death certificates of 648 childhood rhabdomyosarcoma patients. This syndrome is also known as the sarcoma, breast, leukaemia and adrenal gland (SBLA) syndrome.

Sertoli–Leydig cell tumour is a group of tumors composed of variable proportions of Sertoli cells, Leydig cells, and in the case of intermediate and poorly differentiated neoplasms, primitive gonadal stroma and sometimes heterologous elements.
The International Classification of Diseases for Oncology (ICD-O) is a domain-specific extension of the International Statistical Classification of Diseases and Related Health Problems for tumor diseases. This classification is widely used by cancer registries.

Embryonal carcinoma is a relatively uncommon type of germ cell tumour that occurs in the ovaries and testes.

The PAX3 gene encodes a member of the paired box or PAX family of transcription factors. The PAX family consists of nine human (PAX1-PAX9) and nine mouse (Pax1-Pax9) members arranged into four subfamilies. Human PAX3 and mouse Pax3 are present in a subfamily along with the highly homologous human PAX7 and mouse Pax7 genes. The human PAX3 gene is located in the 2q36.1 chromosomal region, and contains 10 exons within a 100 kb region.
Sarcoma botryoides or botryoid sarcoma is a subtype of embryonal rhabdomyosarcoma, that can be observed in the walls of hollow, mucosa lined structures such as the nasopharynx, common bile duct, urinary bladder of infants and young children or the vagina in females, typically younger than age 8. The name comes from the gross appearance of "grape bunches".

Fibroblast growth factor receptor 1 (FGFR1), also known as basic fibroblast growth factor receptor 1, fms-related tyrosine kinase-2 / Pfeiffer syndrome, and CD331, is a receptor tyrosine kinase whose ligands are specific members of the fibroblast growth factor family. FGFR1 has been shown to be associated with Pfeiffer syndrome.
Alveolar rhabdomyosarcoma (ARMS) is a sub-type of the rhabdomyosarcoma soft tissue cancer family whose lineage is from mesenchymal cells and are related to skeletal muscle cells. ARMS tumors resemble the alveoli tissue that can be found in the lungs. Tumor location varies from patient to patient, but is commonly found in the head and neck region, male and female urogenital tracts, the torso, and extremities. Two fusion proteins can be associated with ARMS, but are not necessary, PAX3-FKHR. and PAX7-FKHR. In children and adolescents ARMS accounts for about 1 percent of all malignancies, has an incidence rate of 1 per million, and most cases occur sporadically with no genetic predisposition. PAX3-FOXO1 is now known to drive cancer-promoting gene expression programs through creation of distant genetic elements called super enhancers.
Soft tissue sarcoma refer to a broad group of tumors that originate from connective tissues. They tend to have similar histologic appearance and biological behavior, and can be either benign or malignant. Soft tissue sarcomas can arise in any part of the pet's body but skin and subcutaneous tumors are the most commonly observed. Soft-tissue sarcomas comprise approximately 15% of all skin and subcutaneous tumors in dogs and approximately 7% of all skin and subcutaneous tumors in cats. The variety of different tumors that fall under the category of soft tissue sarcomas includes fibrosarcoma, hemangiopericytoma, liposarcoma, rhabdomyosarcoma, leiomyosarcoma, malignant fibrous histiocytoma, malignant nerve sheath tumors, myxosarcoma, myxofibrosarcoma, mesenchymoma, and spindle cell tumor.

Paired box protein Pax-7 is a protein that in humans is encoded by the PAX7 gene.
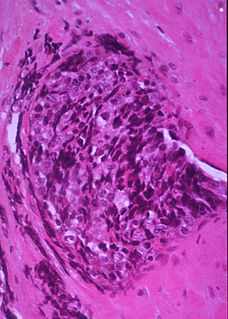
In histopathology, a small-blue-round-cell tumour, also known as a small-round-blue-cell tumor (SRBCT) or a small-round-cell tumour (SRCT), is any one of a group of malignant neoplasms that have a characteristic appearance under the microscope, i.e. consisting of small round cells that stain blue on routine H&E stained sections.
Rhabdomyosarcoma 2 associated transcript (RMST) is a long non-coding RNA. In humans, it is located on chromosome 12q21. It is expressed at higher levels in alveolar rhabdomyosarcoma than in embryonal rhabdomyosarcoma. In the brain, RMST is expressed in the developing ventral midbrain where dopaminergic neurons are formed, the developing isthmus and in the dorsal midline cells of the rostral neural tube. In the midbrain-hindbrain boundary region, its expression is regulated by PAX2.
Sclerosing rhabdomyosarcoma is a rare subtype of rhabdomyosarcoma that was characterized by Folpe et al. in 2002. It is microscopically characterized by primitive round cells forming microalveoli, nests, and cords in a sclerotic background.
A rhabdomyoblast is a cell type which is essential to the diagnosis of a rhabdomyosarcoma. A rhabdomyoblast found histologically is considered diagnostic for embryonal, alveolar, and pleomorphic rhabdomyosarcomas. Histology will show an elongated or round cell, exhibiting an embryonic morphology. Occasionally cells will exhibit cross striations by light microscopy, reflecting sarcomere formation and advancement of differentiation. This differentiated phenotype increases following chemotherapy and radiotherapy.
Childhood rhabdomyosarcoma is a cancer that develops out of the cells that form skeletal muscles. These cells are called rhabdomyoblasts.
Squamous cell carcinomas (SCCs), also known as epidermoid carcinomas, comprise a number of different types of cancer that result from squamous cells. These cells form the surface of the skin and lining of hollow organs in the body and line the respiratory and digestive tracts.

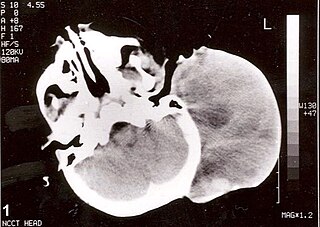
A central nervous system primitive neuroectodermal tumor, often abbreviated as PNET, supratentorial PNET, or CNS-PNET, is one of the 3 types of embryonal central nervous system tumors defined by the World Health Organization. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth. Those cells are usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the spinal cord and cerebrum and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF).