
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

Polyploidy is a condition in which the cells of an organism have more than one pair of (homologous) chromosomes. Most species whose cells have nuclei (eukaryotes) are diploid, meaning they have two complete sets of chromosomes, one from each of two parents; each set contains the same number of chromosomes, and the chromosomes are joined in pairs of homologous chromosomes. However, some organisms are polyploid. Polyploidy is especially common in plants. Most eukaryotes have diploid somatic cells, but produce haploid gametes by meiosis. A monoploid has only one set of chromosomes, and the term is usually only applied to cells or organisms that are normally diploid. Males of bees and other Hymenoptera, for example, are monoploid. Unlike animals, plants and multicellular algae have life cycles with two alternating multicellular generations. The gametophyte generation is haploid, and produces gametes by mitosis; the sporophyte generation is diploid and produces spores by meiosis.

In biology, homology is similarity due to shared ancestry between a pair of structures or genes in different taxa. A common example of homologous structures is the forelimbs of vertebrates, where the wings of bats and birds, the arms of primates, the front flippers of whales, and the forelegs of four-legged vertebrates like dogs and crocodiles are all derived from the same ancestral tetrapod structure. Evolutionary biology explains homologous structures adapted to different purposes as the result of descent with modification from a common ancestor. The term was first applied to biology in a non-evolutionary context by the anatomist Richard Owen in 1843. Homology was later explained by Charles Darwin's theory of evolution in 1859, but had been observed before this, from Aristotle onwards, and it was explicitly analysed by Pierre Belon in 1555.
Molecular evolution describes how inherited DNA and/or RNA change over evolutionary time, and the consequences of this for proteins and other components of cells and organisms. Molecular evolution is the basis of phylogenetic approaches to describing the tree of life. Molecular evolution overlaps with population genetics, especially on shorter timescales. Topics in molecular evolution include the origins of new genes, the genetic nature of complex traits, the genetic basis of adaptation and speciation, the evolution of development, and patterns and processes underlying genomic changes during evolution.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

Comparative genomics is a branch of biological research that examines genome sequences across a spectrum of species, spanning from humans and mice to a diverse array of organisms from bacteria to chimpanzees. This large-scale holistic approach compares two or more genomes to discover the similarities and differences between the genomes and to study the biology of the individual genomes. Comparison of whole genome sequences provides a highly detailed view of how organisms are related to each other at the gene level. By comparing whole genome sequences, researchers gain insights into genetic relationships between organisms and study evolutionary changes. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, Comparative genomics provides a powerful tool for studying evolutionary changes among organisms, helping to identify genes that are conserved or common among species, as well as genes that give unique characteristics of each organism. Moreover, these studies can be performed at different levels of the genomes to obtain multiple perspectives about the organisms.
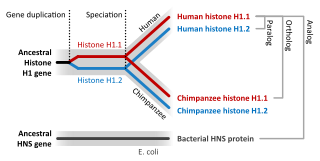
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
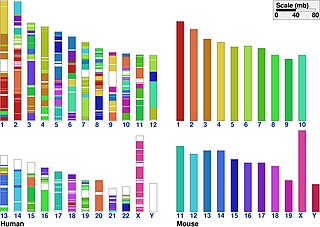
In genetics, the term synteny refers to two related concepts:

Paleopolyploidy is the result of genome duplications which occurred at least several million years ago (MYA). Such an event could either double the genome of a single species (autopolyploidy) or combine those of two species (allopolyploidy). Because of functional redundancy, genes are rapidly silenced or lost from the duplicated genomes. Most paleopolyploids, through evolutionary time, have lost their polyploid status through a process called diploidization, and are currently considered diploids, e.g., baker's yeast, Arabidopsis thaliana, and perhaps humans.

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.
The 2R hypothesis or Ohno's hypothesis, first proposed by Susumu Ohno in 1970, is a hypothesis that the genomes of the early vertebrate lineage underwent two complete genome duplications, and thus modern vertebrate genomes reflect paleopolyploidy. The name derives from the 2 rounds of duplication originally hypothesized by Ohno, but refined in a 1994 version, and the term 2R hypothesis was probably coined in 1999. Variations in the number and timings of genome duplications typically still are referred to as examples of the 2R hypothesis.
Genomic phylostratigraphy is a novel genetic statistical method developed in order to date the origin of specific genes by looking at its homologs across species. It was first developed by Ruđer Bošković Institute in Zagreb, Croatia. The system links genes to their founder gene, allowing us to then determine their age. This could help us better understand many evolutionary processes such as patterns of gene birth throughout evolution, or the relationship between the age of a transcriptome throughout embryonic development. Bioinformatic tools like GenEra have been developed to calculate relative gene ages based on genomic phylostratigraphy.

Gene redundancy is the existence of multiple genes in the genome of an organism that perform the same function. Gene redundancy can result from gene duplication. Such duplication events are responsible for many sets of paralogous genes. When an individual gene in such a set is disrupted by mutation or targeted knockout, there can be little effect on phenotype as a result of gene redundancy, whereas the effect is large for the knockout of a gene with only one copy. Gene knockout is a method utilized in some studies aiming to characterize the maintenance and fitness effects functional overlap.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
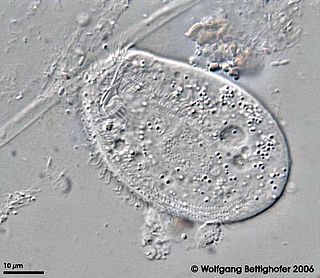
Chilodonella uncinata is a single-celled organism of the ciliate class of alveoles. As a ciliate, C. uncinata has cilia covering its body and a dual nuclear structure, the micronucleus and macronucleus. Unlike some other ciliates, C. uncinata contains millions of minichromosomes in its macronucleus while its micronucleus is estimated to contain 3 chromosomes. Childonella uncinata is the causative agent of Chilodonelloza, a disease that affects the gills and skin of fresh water fish, and may act as a facultative of mosquito larva.
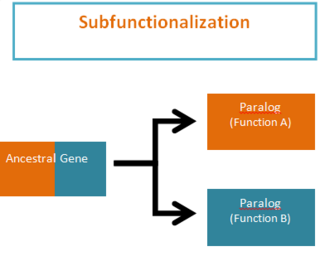
Subfunctionalization was proposed by Stoltzfus (1999) and Force et al. (1999) as one of the possible outcomes of functional divergence that occurs after a gene duplication event, in which pairs of genes that originate from duplication, or paralogs, take on separate functions. Subfunctionalization is a neutral mutation process of constructive neutral evolution; meaning that no new adaptations are formed. During the process of gene duplication paralogs simply undergo a division of labor by retaining different parts (subfunctions) of their original ancestral function. This partitioning event occurs because of segmental gene silencing leading to the formation of paralogs that are no longer duplicates, because each gene only retains a single function. It is important to note that the ancestral gene was capable of performing both functions and the descendant duplicate genes can now only perform one of the original ancestral functions.

Genome evolution is the process by which a genome changes in structure (sequence) or size over time. The study of genome evolution involves multiple fields such as structural analysis of the genome, the study of genomic parasites, gene and ancient genome duplications, polyploidy, and comparative genomics. Genome evolution is a constantly changing and evolving field due to the steadily growing number of sequenced genomes, both prokaryotic and eukaryotic, available to the scientific community and the public at large.
Horizontal or lateral gene transfer is the transmission of portions of genomic DNA between organisms through a process decoupled from vertical inheritance. In the presence of HGT events, different fragments of the genome are the result of different evolutionary histories. This can therefore complicate investigations of the evolutionary relatedness of lineages and species. Also, as HGT can bring into genomes radically different genotypes from distant lineages, or even new genes bearing new functions, it is a major source of phenotypic innovation and a mechanism of niche adaptation. For example, of particular relevance to human health is the lateral transfer of antibiotic resistance and pathogenicity determinants, leading to the emergence of pathogenic lineages.
