
In organic chemistry, a diene ; also diolefin, dy-OH-lə-fin) or alkadiene) is a covalent compound that contains two double bonds, usually among carbon atoms. They thus contain two alkene units, with the standard prefix di of systematic nomenclature. As a subunit of more complex molecules, dienes occur in naturally occurring and synthetic chemicals and are used in organic synthesis. Conjugated dienes are widely used as monomers in the polymer industry. Polyunsaturated fats are of interest to nutrition.

Petrochemicals are the chemical products obtained from petroleum by refining. Some chemical compounds made from petroleum are also obtained from other fossil fuels, such as coal or natural gas, or renewable sources such as maize, palm fruit or sugar cane.

1,3-Butadiene is the organic compound with the formula CH2=CH-CH=CH2. It is a colorless gas that is easily condensed to a liquid. It is important industrially as a precursor to synthetic rubber. The molecule can be viewed as the union of two vinyl groups. It is the simplest conjugated diene.
The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
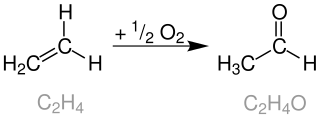
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.

Allylpalladium(II) chloride dimer (APC) is a chemical compound with the formula [(η3-C3H5)PdCl]2. This yellow air-stable compound is an important catalyst used in organic synthesis. It is one of the most widely used transition metal allyl complexes.
In organometallic chemistry, the Green–Davies–Mingos rules predict the regiochemistry for nucleophilic addition to 18-electron metal complexes containing multiple unsaturated ligands. The rules were published in 1978 by organometallic chemists Stephen G. Davies, Malcolm Green, and Michael Mingos. They describe how and where unsaturated hydrocarbon generally become more susceptibile to nucleophilic attack upon complexation.
Telomerization is a reaction that produces a particular kind of oligomer with two distinct end groups. The oligomer is called a telomer. Some telomerizations proceed by radical pathways, many do not. A generic equation is:
Electrophilic substitution of unsaturated silanes involves attack of an electrophile on an allyl- or vinylsilane. An allyl or vinyl group is incorporated at the electrophilic center after loss of the silyl group.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.

Hydrogen auto-transfer, also known as borrowing hydrogen, is the activation of a chemical reaction by temporary transfer of two hydrogen atoms from the reactant to a catalyst and return of those hydrogen atoms back to a reaction intermediate to form the final product. Two major classes of borrowing hydrogen reactions exist: (a) those that result in hydroxyl substitution, and (b) those that result in carbonyl addition. In the former case, alcohol dehydrogenation generates a transient carbonyl compound that is subject to condensation followed by the return of hydrogen. In the latter case, alcohol dehydrogenation is followed by reductive generation of a nucleophile, which triggers carbonyl addition. As borrowing hydrogen processes avoid manipulations otherwise required for discrete alcohol oxidation and the use of stoichiometric organometallic reagents, they typically display high levels of atom-economy and, hence, are viewed as examples of Green chemistry.
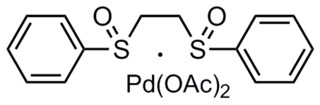
The White catalyst is a transition metal coordination complex named after the chemist by whom it was first synthesized, M. Christina White, a professor at the University of Illinois. The catalyst has been used in a variety of allylic C-H functionalization reactions of α-olefins. In addition, it has been shown to catalyze oxidative Heck reactions.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
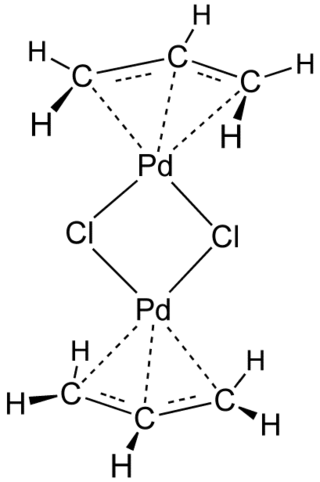
Transition-metal allyl complexes are coordination complexes with allyl and its derivatives as ligands. Allyl is the radical with the connectivity CH2CHCH2, although as a ligand it is usually viewed as an allyl anion CH2=CH−CH2−, which is usually described as two equivalent resonance structures.

In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.
In organic chemistry, hydrovinylation is the formal insertion of an alkene into the C-H bond of ethylene. The more general reaction, hydroalkenylation, is the formal insertion of an alkene into the C-H bond of any terminal alkene. The reaction is catalyzed by metal complexes. A representative reaction is the conversion of styrene and ethylene to 3-phenybutene:
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.
Heterogeneous metal catalyzed cross-coupling is a subset of metal catalyzed cross-coupling in which a heterogeneous metal catalyst is employed. Generally heterogeneous cross-coupling catalysts consist of a metal dispersed on an inorganic surface or bound to a polymeric support with ligands. Heterogeneous catalysts provide potential benefits over homogeneous catalysts in chemical processes in which cross-coupling is commonly employed—particularly in the fine chemical industry—including recyclability and lower metal contamination of reaction products. However, for cross-coupling reactions, heterogeneous metal catalysts can suffer from pitfalls such as poor turnover and poor substrate scope, which have limited their utility in cross-coupling reactions to date relative to homogeneous catalysts. Heterogeneous metal catalyzed cross-couplings, as with homogeneous metal catalyzed ones, most commonly use Pd as the cross-coupling metal.
Copper-catalyzed allylic substitutions are chemical reactions with unique regioselectivity compared to other transition-metal-catalyzed allylic substitutions such as the Tsuji-Trost reaction. They involve copper catalysts and "hard" carbon nucleophiles. The mechanism of copper-catalyzed allylic substitutions involves the coordination of copper to the olefin, oxidative addition and reductive elimination. Enantioselective versions of these reactions have been used in the synthesis of complex molecules, such as (R)-(-)-sporochnol and (S)-(-)-zearalenone.

