Related Research Articles
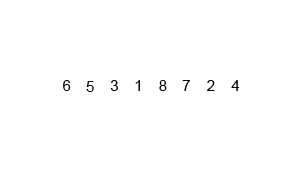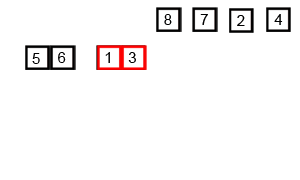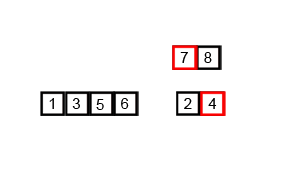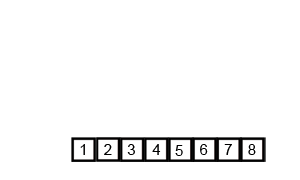
In computer science, merge sort is an efficient, general-purpose, and comparison-based sorting algorithm. Most implementations produce a stable sort, which means that the order of equal elements is the same in the input and output. Merge sort is a divide and conquer algorithm that was invented by John von Neumann in 1945. A detailed description and analysis of bottom-up merge sort appeared in a report by Goldstine and von Neumann as early as 1948.
The Burrows–Wheeler transform rearranges a character string into runs of similar characters. This is useful for compression, since it tends to be easy to compress a string that has runs of repeated characters by techniques such as move-to-front transform and run-length encoding. More importantly, the transformation is reversible, without needing to store any additional data except the position of the first original character. The BWT is thus a "free" method of improving the efficiency of text compression algorithms, costing only some extra computation. The Burrows–Wheeler transform is an algorithm used to prepare data for use with data compression techniques such as bzip2. It was invented by Michael Burrows and David Wheeler in 1994 while Burrows was working at DEC Systems Research Center in Palo Alto, California. It is based on a previously unpublished transformation discovered by Wheeler in 1983. The algorithm can be implemented efficiently using a suffix array thus reaching linear time complexity.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
In bioinformatics, sequence clustering algorithms attempt to group biological sequences that are somehow related. The sequences can be either of genomic, "transcriptomic" (ESTs) or protein origin. For proteins, homologous sequences are typically grouped into families. For EST data, clustering is important to group sequences originating from the same gene before the ESTs are assembled to reconstruct the original mRNA.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Cluster analysis or clustering is the task of grouping a set of objects in such a way that objects in the same group are more similar to each other than to those in other groups (clusters). It is a main task of exploratory data analysis, and a common technique for statistical data analysis, used in many fields, including pattern recognition, image analysis, information retrieval, bioinformatics, data compression, computer graphics and machine learning.
FASTA is a DNA and protein sequence alignment software package first described by David J. Lipman and William R. Pearson in 1985. Its legacy is the FASTA format which is now ubiquitous in bioinformatics.
In statistics, classification is the problem of identifying which of a set of categories (sub-populations) an observation belongs to. Examples are assigning a given email to the "spam" or "non-spam" class, and assigning a diagnosis to a given patient based on observed characteristics of the patient.
Biclustering, block clustering, co-clustering, or two-mode clustering is a data mining technique which allows simultaneous clustering of the rows and columns of a matrix. The term was first introduced by Boris Mirkin to name a technique introduced many years earlier, in 1972, by J. A. Hartigan.
k-means clustering is a method of vector quantization, originally from signal processing, that aims to partition n observations into k clusters in which each observation belongs to the cluster with the nearest mean, serving as a prototype of the cluster. This results in a partitioning of the data space into Voronoi cells. k-means clustering minimizes within-cluster variances, but not regular Euclidean distances, which would be the more difficult Weber problem: the mean optimizes squared errors, whereas only the geometric median minimizes Euclidean distances. For instance, better Euclidean solutions can be found using k-medians and k-medoids.

Fuzzy clustering is a form of clustering in which each data point can belong to more than one cluster.

In electrical engineering and computer science, Lloyd's algorithm, also known as Voronoi iteration or relaxation, is an algorithm named after Stuart P. Lloyd for finding evenly spaced sets of points in subsets of Euclidean spaces and partitions of these subsets into well-shaped and uniformly sized convex cells. Like the closely related k-means clustering algorithm, it repeatedly finds the centroid of each set in the partition and then re-partitions the input according to which of these centroids is closest. In this setting, the mean operation is an integral over a region of space, and the nearest centroid operation results in Voronoi diagrams.
Computational phylogenetics is the application of computational algorithms, methods, and programs to phylogenetic analyses. The goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa. For example, these techniques have been used to explore the family tree of hominid species and the relationships between specific genes shared by many types of organisms.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
In statistics, k-medians clustering is a cluster analysis algorithm. It is a variation of k-means clustering where instead of calculating the mean for each cluster to determine its centroid, one instead calculates the median. This has the effect of minimizing error over all clusters with respect to the 1-norm distance metric, as opposed to the squared 2-norm distance metric
Determining the number of clusters in a data set, a quantity often labelled k as in the k-means algorithm, is a frequent problem in data clustering, and is a distinct issue from the process of actually solving the clustering problem.
An Operational Taxonomic Unit (OTU) is an operational definition used to classify groups of closely related individuals. The term was originally introduced in 1963 by Robert R. Sokal and Peter H. A. Sneath in the context of numerical taxonomy, where an "Operational Taxonomic Unit" is simply the group of organisms currently being studied. In this sense, an OTU is a pragmatic definition to group individuals by similarity, equivalent to but not necessarily in line with classical Linnaean taxonomy or modern evolutionary taxonomy.
CURE is an efficient data clustering algorithm for large databases. Compared with K-means clustering it is more robust to outliers and able to identify clusters having non-spherical shapes and size variances.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
References
- ↑ Edgar, R. C. (2010). "Search and clustering orders of magnitude faster than BLAST". Bioinformatics. 26 (19): 2460–2461. doi: 10.1093/bioinformatics/btq461 . ISSN 1367-4803. PMID 20709691.