Related Research Articles

Cenani–Lenz syndactylism, also known as Cenani–Lenz syndrome or Cenani–syndactylism, is an autosomal recessive congenital malformation syndrome involving both upper and lower extremities.

Ayazi syndrome is a syndrome characterized by choroideremia, congenital deafness and obesity.
Oculocerebrorenal syndrome is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. Lowe syndrome can be considered a cause of Fanconi syndrome.
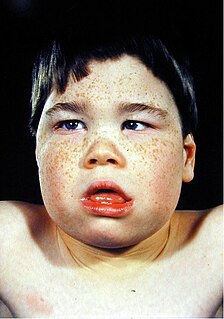
Alpha-thalassemia mental retardation syndrome (ATRX), also called alpha-thalassemia X-linked mental retardation, nondeletion type or ATR-X syndrome, is an X-linked recessive condition associated with a mutation in the ATRX gene. Males with this condition tend to be moderately intellectually disabled and have physical characteristics including coarse facial features, microcephaly, hypertelorism, a depressed nasal bridge, a tented upper lip and an everted lower lip. Mild or moderate anemia, associated with alpha-thalassemia, is part of the condition. Females with this mutated gene have no specific signs or features, but if they do, they may demonstrate skewed X chromosome inactivation.

Kaufman oculocerebrofacial syndrome is an autosomal recessive congenital disorder characterized by mental retardation, brachycephaly, upslanting palpebral fissures, eye abnormalities, and highly arched palate. It was characterized in 1971; eight cases had been identified as of 1995.
Rosselli–Gulienetti syndrome, also known as Zlotogora–Ogur syndrome and Bowen–Armstrong syndrome, is a type of congenital ectodermal dysplasia syndrome. The syndrome is relatively rare and has only been described in a few cases.
Dubowitz syndrome is a rare genetic disorder characterized by microcephaly, stunted growth, and a receding chin. Symptoms vary among patients, but other characteristics include a soft, high-pitched voice, partial webbing of the fingers and toes, palate deformations, genital abnormalities, language difficulties, and an aversion to crowds. The pathogenesis of the disease is yet to be identified, and no medical tests can definitively diagnose the disease. The primary method of diagnosis is to identify facial phenotypes. Since it was first described in 1965 by English physician Victor Dubowitz, over 140 cases have been reported worldwide. Although the majority of cases have been reported from the United States, Germany, and Russia, the disorder appears to affect both genders and all ethnicities equally.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, mental retardation and postnatal growth retardation.

Aristaless related homeobox is a protein that in humans is encoded by the ARX gene.
VLDLR-associated cerebellar hypoplasia (VLDLRCH) is a rare autosomal recessive condition caused by a disruption of the VLDLR gene. First described as a form of cerebral palsy in the 1970s, it is associated with parental consanguinity and is found in secluded communities, with a number of cases described in Hutterite families.

PHD finger protein 8 is a protein that in humans is encoded by the PHF8 gene.
Woodhouse–Sakati syndrome, is a rare autosomal recessive multisystem disorder which causes malformations throughout the body, and deficiencies affecting the endocrine system.
Gerodermia osteodysplastica (GO), is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.
X-linked intellectual disability refers to forms of intellectual disability which are specifically associated with X-linked recessive inheritance.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency. is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Hennekam syndrome also known as intestinal lymphagiectasia–lymphedema–mental retardation syndrome, is an autosomal recessive disorder consisting of intestinal lymphangiectasia, facial anomalies, peripheral lymphedema, and mild to moderate levels of growth and intellectual disability.

Coiled-coil and C2 domain-containing protein 2A that in humans is encoded by the CC2D2A gene.
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem. Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH.
Wiedemann–Rautenstrauch (WR) syndrome, also known as neonatal progeroid syndrome, is a rare autosomal recessive progeroid syndrome. There have been over 30 cases of WR. WR is associated with abnormalities in bone maturation, and lipids and hormone metabolism.
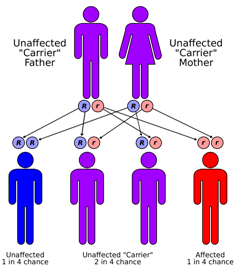
Sanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia, but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere. The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
References
- ↑ Warkany J (1961). "Intrauterine growth retardation". Am. J. Dis. Child. 102 (2): 249–79. doi:10.1001/archpedi.1961.02080010251018. PMID 13783175.
- ↑ Lubs H (1999). "XLMR genes: update 1998". Am. J. Med. Genet. 83 (4): 237–47. doi:10.1002/(SICI)1096-8628(19990402)83:4<237::AID-AJMG2>3.0.CO;2-8. PMID 10208155.
- ↑ Chiurazzi P (2008). "XLMR genes: update 2007". Eur. J. Hum. Genet. 16 (4): 422–34. doi: 10.1038/sj.ejhg.5201994 . PMID 18197188.