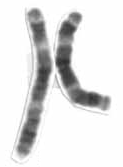
Smith–Magenis Syndrome (SMS), also known as 17p- syndrome, is a microdeletion syndrome characterized by an abnormality in the short (p) arm of chromosome 17. It has features including intellectual disability, facial abnormalities, difficulty sleeping, and numerous behavioral problems such as self-harm. Smith–Magenis syndrome affects an estimated between 1 in 15,000 to 1 in 25,000 individuals.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

2q37 deletion syndrome is a disorder caused by the deletion of a small piece of chromosome 2 in which one or more of 3 sub-bands, 2q37.1, 2q37.2, and 2q37.3, of the last band of one of the chromosome 2’s long arms are deleted. The first report of this disorder was in 1989.
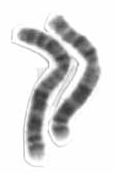
2p15-16.1 microdeletion is an extremely rare genetic disorder caused by a small deletion in the short arm of human chromosome 2. First described in two patients in 2007, by 2013 only 21 people have been reported as having the disorder in the medical literature.
Potocki–Lupski syndrome (PTLS), also known as dup(17)p11.2p11.2 syndrome, trisomy 17p11.2 or duplication 17p11.2 syndrome, is a contiguous gene syndrome involving the microduplication of band 11.2 on the short arm of human chromosome 17 (17p11.2). The duplication was first described as a case study in 1996. In 2000, the first study of the disease was released, and in 2007, enough patients had been gathered to complete a comprehensive study and give it a detailed clinical description. PTLS is named for two researchers involved in the latter phases, Drs. Lorraine Potocki and James R. Lupski of Baylor College of Medicine.

Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.
Koolen–De Vries syndrome (KdVS), also known as 17q21.31 microdeletion syndrome, is a rare genetic disorder caused by a deletion of a segment of chromosome 17 which contains six genes. This deletion syndrome was discovered independently in 2006 by three different research groups.
DECIPHER is a web-based resource and database of genomic variation data from analysis of patient DNA. It documents submicroscopic chromosome abnormalities and pathogenic sequence variants, from over 25000 patients and maps them to the human genome using Ensembl or UCSC Genome Browser. In addition it catalogues the clinical characteristics from each patient and maintains a database of microdeletion/duplication syndromes, together with links to relevant scientific reports and support groups.
Non-allelic homologous recombination (NAHR) is a form of homologous recombination that occurs between two lengths of DNA that have high sequence similarity, but are not alleles.
esophageal candidiasis1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.
1q21.1 duplication syndrome or 1q21.1 (recurrent) microduplication is a rare aberration of chromosome 1.
2q37 monosomy is a rare genetic disorder caused by a deletion of a segment at the end of chromosome 2.
22q11.2 duplication syndrome is a rare genetic disorder caused by a duplication of a segment at the end of chromosome 22.

Distal 18q- is a genetic condition caused by a deletion of genetic material within one of the two copies of chromosome 18. The deletion involves the distal section of 18q and typically extends to the tip of the long arm of chromosome 18.
Chromosomal deletion syndromes result from deletion of parts of chromosomes. Depending on the location, size, and whom the deletion is inherited from, there are a few known different variations of chromosome deletions. Chromosomal deletion syndromes typically involve larger deletions that are visible using karyotyping techniques. Smaller deletions result in Microdeletion syndrome, which are detected using fluorescence in situ hybridization (FISH)
Burnside–Butler syndrome is a name that has been applied to the effects of microdeletion of DNA sequences involving four neurodevelopmental genes. Varying developmental and psychiatric disorders have been attributed to the microdeletion; however, the great majority of people with the deletion do not have any clinical features associated with it. More studies are needed to delineate the range of clinical presentation.

Ankyrin repeat domain 11 is a protein that in humans is encoded by the ANKRD11 gene.

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
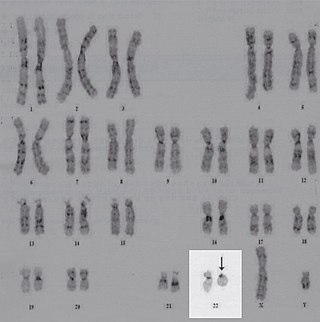
Ring chromosome 22, also known as ring 22, is a rare chromosomal disorder. Ring chromosomes occur when the ends of a chromosome lose material and fuse into a ring shape; in the case of ring 22, this occurs for chromosome 22, the last numbered human autosome. Ring chromosome 22 is marked by a number of consistent traits, such as intellectual disability, speech delay, hypotonia, and hyperactivity. The condition has a similar phenotype to Phelan-McDermid syndrome, as the loss of the SHANK3 gene is implicated in both.

DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22. While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental delay, intellectual disability and cleft palate. Associated conditions include kidney problems, schizophrenia, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.