
Jaundice, also known as icterus, is a yellowish or greenish pigmentation of the skin and sclera due to high bilirubin levels. Jaundice in adults is typically a sign indicating the presence of underlying diseases involving abnormal heme metabolism, liver dysfunction, or biliary-tract obstruction. The prevalence of jaundice in adults is rare, while jaundice in babies is common, with an estimated 80% affected during their first week of life. The most commonly associated symptoms of jaundice are itchiness, pale feces, and dark urine.

Ascites is the abnormal build-up of fluid in the abdomen. Technically, it is more than 25 ml of fluid in the peritoneal cavity, although volumes greater than one liter may occur. Symptoms may include increased abdominal size, increased weight, abdominal discomfort, and shortness of breath. Complications can include spontaneous bacterial peritonitis.
Liver function tests, also referred to as a hepatic panel, are groups of blood tests that provide information about the state of a patient's liver. These tests include prothrombin time (PT/INR), activated partial thromboplastin time (aPTT), albumin, bilirubin, and others. The liver transaminases aspartate transaminase and alanine transaminase are useful biomarkers of liver injury in a patient with some degree of intact liver function.

Gilbert syndrome (GS) is a syndrome in which the liver of affected individuals processes bilirubin more slowly than the majority. Many people never have symptoms. Occasionally jaundice may occur.

Budd–Chiari syndrome is a very rare condition, affecting one in a million adults. The condition is caused by occlusion of the hepatic veins that drain the liver. The symptoms are non-specific and vary widely, but it may present with the classical triad of abdominal pain, ascites, and liver enlargement. It is usually seen in younger adults, with the median age at diagnosis between the ages of 35 and 40, and it has a similar incidence in males and females. The syndrome can be fulminant, acute, chronic, or asymptomatic. Subacute presentation is the most common form.

Alcoholic hepatitis is hepatitis due to excessive intake of alcohol. Patients typically have a history of at least 10 years of heavy alcohol intake, typically 8-10 drinks per day. It is usually found in association with fatty liver, an early stage of alcoholic liver disease, and may contribute to the progression of fibrosis, leading to cirrhosis. Symptoms may present acutely after a large amount of alcoholic intake in a short time period, or after years of excess alcohol intake. Signs and symptoms of alcoholic hepatitis include jaundice, ascites, fatigue and hepatic encephalopathy. Mild cases are self-limiting, but severe cases have a high risk of death. Severe cases may be treated with glucocorticoids. The condition often comes on suddenly and may progress in severity very rapidly.

Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is an autoimmune disease of the liver. It results from a slow, progressive destruction of the small bile ducts of the liver, causing bile and other toxins to build up in the liver, a condition called cholestasis. Further slow damage to the liver tissue can lead to scarring, fibrosis, and eventually cirrhosis.

Alagille syndrome (ALGS) is a genetic disorder that affects primarily the liver and the heart. Problems associated with the disorder generally become evident in infancy or early childhood. The disorder is inherited in an autosomal dominant pattern, and the estimated prevalence of Alagille syndrome is 1 in every 30,000 to 1 in every 40,000 live births. It is named after the French pediatrician Daniel Alagille, who first described the condition in 1969.
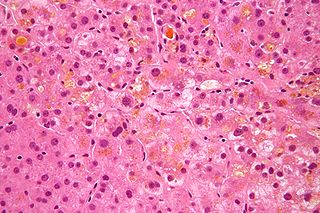
Cholestasis is a condition where the flow of bile from the liver to the duodenum is impaired. The two basic distinctions are:

Exocrine pancreatic insufficiency (EPI) is the inability to properly digest food due to a lack or reduction of digestive enzymes made by the pancreas. EPI can occur in humans and is prevalent in many conditions such as cystic fibrosis, Shwachman–Diamond syndrome, different types of pancreatitis, multiple types of diabetes mellitus, advanced renal disease, older adults, celiac disease, IBS-D, IBD, HIV, alcohol-related liver disease, Sjogren syndrome, tobacco use, and use of somatostatin analogues.
Progressive familial intrahepatic cholestasis (PFIC) is a group of familial cholestatic conditions caused by defects in biliary epithelial transporters. The clinical presentation usually occurs first in childhood with progressive cholestasis. This usually leads to failure to thrive, cirrhosis, and the need for liver transplantation.
Neonatal cholestasis refers to elevated levels of conjugated bilirubin identified in newborn infants within the first few months of life. Conjugated hyperbilirubinemia is clinically defined as >20% of total serum bilirubin or conjugated bilirubin concentration greater than 1.0 mg/dL regardless of total serum bilirubin concentration. The differential diagnosis for neonatal cholestasis can vary extensively. However, the underlying disease pathology is caused by improper transport and/or defects in excretion of bile from hepatocytes leading to an accumulation of conjugated bilirubin in the body. Generally, symptoms associated with neonatal cholestasis can vary based on the underlying cause of the disease. However, most infants affected will present with jaundice, scleral icterus, failure to thrive, acholic or pale stools, and dark urine.
Intrahepatic cholestasis of pregnancy (ICP), also known as obstetric cholestasis, cholestasis of pregnancy, jaundice of pregnancy, and prurigo gravidarum, is a medical condition in which cholestasis occurs during pregnancy. It typically presents with itching and can lead to complications for both mother and fetus.
Hy's law is a rule of thumb that a patient is at high risk of a fatal drug-induced liver injury if given a medication that causes hepatocellular injury with jaundice. The law is based on observations by Hy Zimmerman, a major scholar of drug-induced liver injury. Some have suggested the principle be called a hypothesis or observation.

Elevated alkaline phosphatase occurs when levels of alkaline phosphatase (ALP) exceed the reference range. This group of enzymes has a low substrate specificity and catalyzes the hydrolysis of phosphate esters in a basic environment. The major function of alkaline phosphatase is transporting chemicals across cell membranes. Alkaline phosphatases are present in many human tissues, including bone, intestine, kidney, liver, placenta and white blood cells. Damage to these tissues causes the release of ALP into the bloodstream. Elevated levels can be detected through a blood test. Elevated alkaline phosphate is associated with certain medical conditions or syndromes. It serves as a significant indicator for certain medical conditions, diseases and syndromes.
Cholestatic pruritus is the sensation of itch due to nearly any liver disease, but the most commonly associated entities are primary biliary cholangitis, primary sclerosing cholangitis, obstructive choledocholithiasis, carcinoma of the bile duct, cholestasis, and chronic hepatitis C viral infection and other forms of viral hepatitis.
A liver support system or diachysis is a type of therapeutic device to assist in performing the functions of the liver. Such systems focus either on removing the accumulating toxins, or providing additional replacement of the metabolic functions of the liver through the inclusion of hepatocytes to the device. This system is in trial to help people with acute liver failure (ALF) or acute-on-chronic liver failure.

Bilirubin glucuronide is a water-soluble reaction intermediate over the process of conjugation of indirect bilirubin. Bilirubin glucuronide itself belongs to the category of conjugated bilirubin along with bilirubin di-glucuronide. However, only the latter one is primarily excreted into the bile in the normal setting.

Odevixibat, sold under the brand name Bylvay, is a medication for the treatment of progressive familial intrahepatic cholestasis. It is taken by mouth. Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT). It was developed by Albireo Pharma.
Hyperbilirubinemia is a clinical condition describing an elevation of blood bilirubin level due to the inability to properly metabolise or excrete bilirubin, a product of erythrocytes breakdown. In severe cases, it is manifested as jaundice, the yellowing of tissues like skin and the sclera when excess bilirubin deposits in them. The US records 52,500 jaundice patients annually. By definition, bilirubin concentration of greater than 3 mg/ml is considered hyperbilirubinemia, following which jaundice progressively develops and becomes apparent when plasma levels reach 20 mg/ml. Rather than a disease itself, hyperbilirubinemia is indicative of multifactorial underlying disorders that trace back to deviations from regular bilirubin metabolism. Diagnosis of hyperbilirubinemia depends on physical examination, urinalysis, serum tests, medical history and imaging to identify the cause. Genetic diseases, alcohol, pregnancy and hepatitis viruses affect the likelihood of hyperbilirubinemia. Causes of hyperbilirubinemia mainly arise from the liver. These include haemolytic anaemias, enzymatic disorders, liver damage and gallstones. Hyperbilirubinemia itself is often benign. Only in extreme cases does kernicterus, a type of brain injury, occur. Therapy for adult hyperbilirubinemia targets the underlying diseases but patients with jaundice often have poor outcomes.
