Inclusion body myositis (IBM) is the most common inflammatory muscle disease in older adults. The disease is characterized by slowly progressive weakness and wasting of both proximal muscles and distal muscles, most apparent in the finger flexors and knee extensors. IBM is often confused with an entirely different class of diseases, called hereditary inclusion body myopathies (hIBM). The "M" in hIBM is an abbreviation for "myopathy" while the "M" in IBM is for "myositis". In IBM, two processes appear to occur in the muscles in parallel, one autoimmune and the other degenerative. Inflammation is evident from the invasion of muscle fibers by immune cells. Degeneration is characterized by the appearance of holes, deposits of abnormal proteins, and filamentous inclusions in the muscle fibers. sIBM is a rare disease, with a prevalence ranging from 1 to 71 individuals per million.

Myalgia is the medical term for muscle pain. Myalgia is a symptom of many diseases. The most common cause of acute myalgia is the overuse of a muscle or group of muscles; another likely cause is viral infection, especially when there has been no trauma.

Adenosine monophosphate deaminase deficiency type 1 or AMPD1, is a human metabolic disorder in which the body consistently lacks the enzyme AMP deaminase, in sufficient quantities. This may result in exercise intolerance, muscle pain and muscle cramping. The disease was formerly known as myoadenylate deaminase deficiency (MADD).

Benign fasciculation syndrome (BFS) is characterized by fasciculation (twitching) of voluntary muscles in the body. The twitching can occur in any voluntary muscle group but is most common in the eyelids, arms, hands, fingers, legs, and feet. The tongue can also be affected. The twitching may be occasional to continuous. BFS must be distinguished from other conditions that include muscle twitches.

Glycogen storage disease type V, also known as McArdle's disease, is a metabolic disorder, one of the metabolic myopathies, more specifically a muscle glycogen storage disease, caused by a deficiency of myophosphorylase. Its incidence is reported as one in 100,000, roughly the same as glycogen storage disease type I.
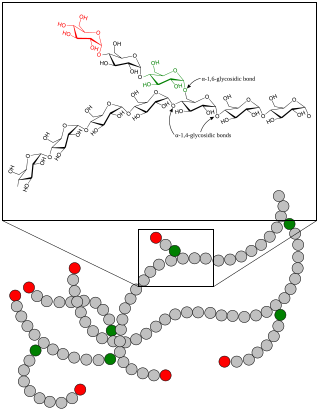
A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Rhabdomyolysis is a condition in which damaged skeletal muscle breaks down rapidly, often due to high intensity exercise over a short period of time. Symptoms may include muscle pains, weakness, vomiting, and confusion. There may be tea-colored urine or an irregular heartbeat. Some of the muscle breakdown products, such as the protein myoglobin, are harmful to the kidneys and can cause acute kidney injury.

Phosphofructokinase deficiency is a rare muscular metabolic disorder, with an autosomal recessive inheritance pattern.
In medicine, myopathy is a disease of the muscle in which the muscle fibers do not function properly. Myopathy means muscle disease. This meaning implies that the primary defect is within the muscle, as opposed to the nerves or elsewhere.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

Myositis is a rarely-encountered medical condition characterized by inflammation affecting the muscles. The manifestations of this condition may include skin issues, muscle weakness, and the potential involvement of other organs. Additionally, systemic symptoms like weight loss, fatigue, and low-grade fever can manifest in individuals with myositis.

Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.

Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in the center of muscle cells instead of their normal location at the periphery.

Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.
Thyrotoxic myopathy (TM) is a neuromuscular disorder that develops due to the overproduction of the thyroid hormone thyroxine. Also known as hyperthyroid myopathy, TM is one of many myopathies that lead to muscle weakness and muscle tissue breakdown. Evidence indicates the onset may be caused by hyperthyroidism. Physical symptoms of TM may include muscle weakness, the breakdown of muscle tissue, fatigue, and heat intolerance. Physical acts such as lifting objects and climbing stairs may become increasingly difficult. If untreated, TM can be an extremely debilitating disorder that can, in extreme rare cases, lead to death. If diagnosed and treated properly the effects can be controlled and in most cases reversed leaving no lasting effects.
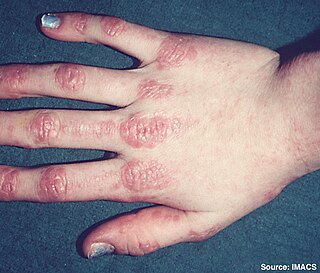
Inflammatory myopathy, also known as idiopathic inflammatory myopathy (IIM), is disease featuring muscle weakness, inflammation of muscles (myositis), and in some types, muscle pain. The cause of much inflammatory myopathy is unknown (idiopathic), and such cases are classified according to their symptoms and signs, electromyography, MRI, and laboratory findings. It can also be associated with underlying cancer. The main classes of idiopathic inflammatory myopathy are polymyositis (PM), dermatomyositis (DM), inclusion-body myositis (IBM), immune-mediated necrotising myopathy (IMNM), and focal autoimmune myositis.

Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects that interfere with muscle's ability to create energy, causing a low ATP reservoir within the muscle cell.
Brody myopathy, also called Brody disease, is a rare disorder that affects skeletal muscle function. BD was first characterized in 1969 by Dr. Irwin A. Brody at Duke University Medical Center. Individuals with BD have difficulty relaxing their muscles after exercise. This difficulty in relaxation leads to symptoms including cramps, stiffness, and discomfort in the muscles of the limbs and face. Symptoms are heightened by exercise and commonly progress in severity throughout adulthood.
Statin-associated autoimmune myopathy (SAAM), also known as anti-HMGCR myopathy, is a very rare form of muscle damage caused by the immune system in people who take statin medications. However, there are cases of SAAM in patients who have not taken statin medication, and this can be explained by the exposure to natural sources of statin such as red yeast rice, which is statin rich. This theory is supported by the higher prevalence of statin-naive SAAM patients in Asian cohorts, who have statin-rich diets.
Autophagic vacuolar myopathy (AVM) consists of multiple rare genetic disorders with common histological and pathological features on muscle biopsy. The features highlighted are vacuolar membranes of the autophagic vacuoles having sarcolemmal characteristics and an excess of autophagic vacuoles. There are currently five types of AVM identified. The signs and symptoms become more severe over the course of the disease. It begins with an inability to pick up small objects and progresses to difficulty in walking. The age of onset varies from early childhood to late adulthood, affecting people of all ages.




