Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
The aldol reaction is a reaction that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
The Robinson annulation is a chemical reaction used in organic chemistry for ring formation. It was discovered by Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds. The method uses a ketone and a methyl vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring by a Michael addition followed by an aldol condensation. This procedure is one of the key methods to form fused ring systems.

The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which a prochiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.

Biocatalysis refers to the use of living (biological) systems or their parts to speed up (catalyze) chemical reactions. In biocatalytic processes, natural catalysts, such as enzymes, perform chemical transformations on organic compounds. Both enzymes that have been more or less isolated and enzymes still residing inside living cells are employed for this task. Modern biotechnology, specifically directed evolution, has made the production of modified or non-natural enzymes possible. This has enabled the development of enzymes that can catalyze novel small molecule transformations that may be difficult or impossible using classical synthetic organic chemistry. Utilizing natural or modified enzymes to perform organic synthesis is termed chemoenzymatic synthesis; the reactions performed by the enzyme are classified as chemoenzymatic reactions.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.
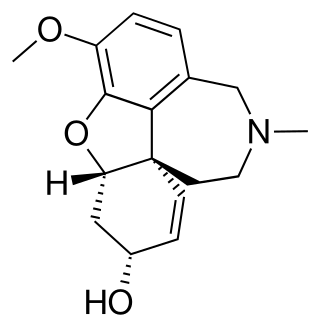
The article concerns the total synthesis of galanthamine, a drug used for the treatment of mild to moderate Alzheimer's disease.
In chemistry, the Noyori asymmetric hydrogenation refers to methodology for enantioselective reduction of ketones and related functional groups. This methodology was introduced by Ryoji Noyori, who shared the Nobel Prize in Chemistry in 2001 for contributions to asymmetric hydrogenation. These hydrogenations are used in the production of several drugs, such as the antibacterial levofloxin, the antibiotic carbapenem, and the antipsychotic agent BMS181100.

Danishefsky's diene is an organosilicon compound and a diene with the formal name trans-1-methoxy-3-trimethylsilyloxy-buta-1,3-diene named after Samuel J. Danishefsky. Because the diene is very electron-rich it is a very reactive reagent in Diels-Alder reactions. This diene reacts rapidly with electrophilic alkenes, such as maleic anhydride. The methoxy group promotes highly regioselective additions. The diene is known to react with amines, aldehydes, alkenes and alkynes. Reactions with imines and nitro-olefins have been reported.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
In Lewis acid catalysis of organic reactions, a metal-based Lewis acid acts as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.
In organic chemistry, the Roskamp reaction is a name reaction describing the reaction between α-diazoesters (such as ethyl diazoacetate) and aldehydes to form β-ketoesters, often utilizing various Lewis acids (such as BF3, SnCl2, and GeCl2) as catalysts. The reaction is notable for its mild reaction conditions and selectivity.
F. Dean Toste is the Gerald E. K. Branch Distinguished Professor of Chemistry at the University of California, Berkeley and faculty scientist at the chemical sciences division of Lawrence Berkeley National Lab. He is a prominent figure in the field of organic chemistry and is best known for his contributions to gold chemistry and asymmetric ion-pairing catalysis. Toste was elected a member of the National Academy of Sciences in 2020, and a member of the American Academy of Arts and Sciences in 2018.
In organic chemistry, the Conia-ene reaction is an intramolecular cyclization reaction between an enolizable carbonyl such as an ester or ketone and an alkyne or alkene, giving a cyclic product with a new carbon-carbon bond. As initially reported by J. M. Conia and P. Le Perchec, the Conia-ene reaction is a heteroatom analog of the ene reaction that uses an enol as the ene component. Like other pericyclic reactions, the original Conia-ene reaction required high temperatures to proceed, limiting its wider application. However, subsequent improvements, particularly in metal catalysis, have led to significant expansion of reaction scope. Consequently, various forms of the Conia-ene reaction have been employed in the synthesis of complex molecules and natural products.