Related Research Articles

Vitamin K refers to structurally similar, fat-soluble vitamers found in foods and marketed as dietary supplements. The human body requires vitamin K for post-synthesis modification of certain proteins that are required for blood coagulation or for controlling binding of calcium in bones and other tissues. The complete synthesis involves final modification of these so-called "Gla proteins" by the enzyme gamma-glutamyl carboxylase that uses vitamin K as a cofactor.

Biotin, also called vitamin B7, is one of the B vitamins. It is involved in a wide range of metabolic processes, both in humans and in other organisms, primarily related to the utilization of fats, carbohydrates, and amino acids. The name biotin derives from the Greek word “bios” (to live) and the suffix “-in” (a general chemical suffix used in organic chemistry).
A congenital disorder of glycosylation is one of several rare inborn errors of metabolism in which glycosylation of a variety of tissue proteins and/or lipids is deficient or defective. Congenital disorders of glycosylation are sometimes known as CDG syndromes. They often cause serious, sometimes fatal, malfunction of several different organ systems in affected infants. The most common sub-type is PMM2-CDG where the genetic defect leads to the loss of phosphomannomutase 2 (PMM2), the enzyme responsible for the conversion of mannose-6-phosphate into mannose-1-phosphate.

Ornithine transcarbamylase (OTC) is an enzyme that catalyzes the reaction between carbamoyl phosphate (CP) and ornithine (Orn) to form citrulline (Cit) and phosphate (Pi). There are two classes of OTC: anabolic and catabolic. This article focuses on anabolic OTC. Anabolic OTC facilitates the sixth step in the biosynthesis of the amino acid arginine in prokaryotes. In contrast, mammalian OTC plays an essential role in the urea cycle, the purpose of which is to capture toxic ammonia and transform it into urea, a less toxic nitrogen source, for excretion.

Methylmalonic acidemia, also called methylmalonic aciduria, is an autosomal recessive metabolic disorder that disrupts normal amino acid metabolism. It is a classical type of organic acidemia. The result of this condition is the inability to properly digest specific fats and proteins, which in turn leads to a buildup of a toxic level of methylmalonic acid in the blood.
Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency, is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.
Holocarboxylase synthetase deficiency is an inherited metabolic disorder in which the body is unable to use the vitamin biotin effectively. This disorder is classified as a multiple carboxylase deficiency, a group of disorders characterized by impaired activity of certain enzymes that depend on biotin. Symptoms are very similar to biotinidase deficiency and treatment – large doses of biotin – is also the same.

Pyruvate carboxylase (PC) encoded by the gene PC is an enzyme of the ligase class that catalyzes the physiologically irreversible carboxylation of pyruvate to form oxaloacetate (OAA).
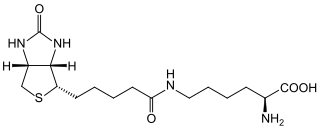
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.

Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.
HLCS, also known as protein–biotin ligase, is a family of enzymes. This enzyme is important for the effective use of biotin, a B vitamin found in foods such as liver, egg yolks, and milk. In many of the body's tissues, holocarboxylase synthetase activates other specific enzymes by attaching biotin to them. These carboxylases are involved in many critical cellular functions, including the production and breakdown of proteins, fats, and carbohydrates.

3-Methylcrotonyl-CoA carboxylase deficiency also known as 3-Methylcrotonylglycinuria or BMCC deficiency is an inherited disorder in which the body is unable to process certain proteins properly. People with this disorder have inadequate levels of an enzyme that helps break down proteins containing the amino acid leucine. This condition affects an estimated 1 in 50,000 individuals worldwide.
Pyruvate carboxylase deficiency is an inherited disorder that causes lactic acid to accumulate in the blood. High levels of these substances can damage the body's organs and tissues, particularly in the nervous system. Pyruvate carboxylase deficiency is a rare condition, with an estimated incidence of 1 in 250,000 births worldwide. Type A of the disease appears to be much more common in some Algonkian Indian tribes in eastern Canada, while the type B disease is more present in European populations.

Sucrose intolerance or genetic sucrase-isomaltase deficiency (GSID) is the condition in which sucrase-isomaltase, an enzyme needed for proper metabolism of sucrose (sugar) and starch, is not produced or the enzyme produced is either partially functional or non-functional in the small intestine. All GSID patients lack fully functional sucrase, while the isomaltase activity can vary from minimal functionality to almost normal activity. The presence of residual isomaltase activity may explain why some GSID patients are better able to tolerate starch in their diet than others with GSID.

Propionyl-CoA carboxylase (PCC) catalyses the carboxylation reaction of propionyl-CoA in the mitochondrial matrix. PCC has been classified both as a ligase and a lyase. The enzyme is biotin-dependent. The product of the reaction is (S)-methylmalonyl CoA.
Methylcrotonyl CoA carboxylase (MCC) is a biotin-requiring enzyme located in the mitochondria. MCC uses bicarbonate as a carboxyl group source to catalyze the carboxylation of a carbon adjacent to a carbonyl group performing the fourth step in processing leucine, an essential amino acid.

Gamma-glutamyl carboxylase is an enzyme that in humans is encoded by the GGCX gene, located on chromosome 2 at 2p12.
Biotin deficiency is a nutritional disorder which can become serious, even fatal, if allowed to progress untreated. It can occur in people of any age, ancestry, or gender. Biotin is part of the B vitamin family. Biotin deficiency rarely occurs among healthy people because the daily requirement of biotin is low, many foods provide adequate amounts of it, intestinal bacteria synthesize small amounts of it, and the body effectively scavenges and recycles it in the kidneys during production of urine. However, deficiencies can be caused by consuming raw egg whites over a period of weeks to months. Egg whites contain high levels of avidin, a protein that binds biotin strongly. When cooked, avidin is partially denatured and binding to biotin is reduced. However one study showed that 30-40% of the avidin activity was still present in the white after frying or boiling. Genetic disorders such as biotinidase deficiency, multiple carboxylase deficiency, and holocarboxylase synthetase deficiency can also lead to inborn or late-onset forms of biotin deficiency. In all cases – dietary, genetic, or otherwise – supplementation with biotin is the primary method of treatment.
Multiple carboxylase deficiency is a form of metabolic disorder involving failures of carboxylation enzymes.
References
- ↑ "ENZYME - 3.5.1.12 Biotinidase". enzyme.expasy.org. Retrieved 2022-03-06.
- ↑ Pindolia, K; Jordan, M; Wolf, B (September 2010). "Analysis of mutations causing biotinidase deficiency". Human Mutation. 31 (9): 983–91. doi: 10.1002/humu.21303 . PMID 20556795. S2CID 26622938.
- Dobrowolski SF, Angeletti J, Banas RA, Naylor EW (2003). "Real time PCR assays to detect common mutations in the biotinidase gene and application of mutational analysis to newborn screening for biotinidase deficiency". Mol Genet Metab. 78 (2): 100–7. doi:10.1016/S1096-7192(02)00231-7. PMID 12618081.
- McMahon RJ (2002). "Biotin in metabolism and molecular biology". Annu Rev Nutr. 22: 221–39. doi:10.1146/annurev.nutr.22.121101.112819. PMID 12055344.
- Neto EC, Schulte J, Rubim R, Lewis E, DeMari J, Castilhos C, Brites A, Giugliani R, Jensen KP, Wolf B (2004). "Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations". Braz J Med Biol Res. 37 (3): 295–9. doi: 10.1590/S0100-879X2004000300001 . PMID 15060693.
- Weber P, Scholl S, Baumgartner ER (2004). "Outcome in patients with profound biotinidase deficiency: relevance of newborn screening". Dev Med Child Neurol. 46 (7): 481–4. doi: 10.1111/j.1469-8749.2004.tb00509.x . PMID 15230462. S2CID 23111366.
- Hymes J, Stanley CM, Wolf B (2001). "Mutations in BTD causing biotinidase deficiency". Hum Mutat. 18 (5): 375–81. doi:10.1002/humu.1208. PMID 11668630. S2CID 31031088.
- Wolf B (2003). "Biotinidase Deficiency: New Directions and Practical Concerns". Curr Treat Options Neurol. 5 (4): 321–328. doi:10.1007/s11940-003-0038-4. PMID 12791199. S2CID 25158348.
![]() This article incorporates public domain material from the United States National Library of Medicine document: "Genetics Home Reference".
This article incorporates public domain material from the United States National Library of Medicine document: "Genetics Home Reference".