Related Research Articles

Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others. Since the development of methods of high-throughput production of gene and protein sequences, the rate of addition of new sequences to the databases increased exponentially. Such a collection of sequences does not, by itself, increase the scientist's understanding of the biology of organisms. However, comparing these new sequences to those with known functions is a key way of understanding the biology of an organism from which the new sequence comes. Thus, sequence analysis can be used to assign function to genes and proteins by the study of the similarities between the compared sequences. Nowadays, there are many tools and techniques that provide the sequence comparisons and analyze the alignment product to understand its biology.
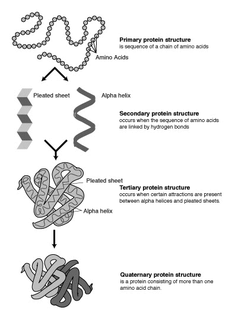
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its folding and its secondary and tertiary structure from its primary structure. Structure prediction is fundamentally different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by bioinformatics and theoretical chemistry; it is highly important in medicine and biotechnology.
Critical Assessment of protein Structure Prediction, or CASP, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence, many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, and binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. Structural bioinformatics main objectives are the creation of new methods to deal with biological macromolecules data to solve problems in biology and generate new knowledge.
Rosetta@home is a distributed computing project for protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker laboratory at the University of Washington. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
The global distance test (GDT), also written as GDT_TS to represent "total score", is a measure of similarity between two protein structures with known amino acid correspondences but different tertiary structures. It is most commonly used to compare the results of protein structure prediction to the experimentally determined structure as measured by X-ray crystallography or protein NMR. The GDT metric as described by its author Adam Zemla is intended as a more accurate measurement than the more common RMSD metric, which is sensitive to outlier regions created for example by poor modeling of individual loop regions in a structure that is otherwise reasonably accurate. GDT_TS measurements are used as major assessment criteria in the production of results from the Critical Assessment of Structure Prediction (CASP), a large-scale experiment in the structure prediction community dedicated to assessing current modeling techniques and identifying their primary deficiencies. In general, the higher GDT_TS is, the better a given model is in comparison to reference structure.

David Baker is an American biochemist and computational biologist who has pioneered methods to predict and design the three-dimensional structures of proteins. He is the Henrietta and Aubrey Davis Endowed Professor in Biochemistry and an adjunct professor of Genome Sciences, Bioengineering, Chemical Engineering, Computer Science, and Physics at the University of Washington. He serves as the Director of the Rosetta Commons, a consortium of labs and researchers that develop biomolecular structure prediction and design software. He is a Howard Hughes Medical Institute investigator and a member of the United States National Academy of Sciences. He is also the director of the University of Washington's Institute for Protein Design.
3D-Jury is a metaserver that aggregates and compares models from various protein structure prediction servers. It takes in groups of predictions made by a collection of servers and assigns each pair a 3D-Jury score, based on structural similarity. The score is generated by counting the number of Cα atoms in the two predictions within 3.5 Å of each other after being superpositioned. To improve accuracy of the final model, users can select the prediction servers from which to aggregate results.

RAPTOR is protein threading software used for protein structure prediction. It has been replaced by RaptorX, which is much more accurate than RAPTOR.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.
SWISS-MODEL is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. Homology modeling is currently the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures ("templates") to build models for evolutionary related proteins ("targets").
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.

Burkhard Rost is a scientist leading the Department for Computational Biology & Bioinformatics at the Faculty of Informatics of the Technical University of Munich (TUM). Rost chairs the Study Section Bioinformatics Munich involving the TUM and the Ludwig Maximilian University of Munich (LMU) in Munich. From 2007-2014 Rost was President of the International Society for Computational Biology (ISCB).

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
Rita Casadio is a Professor of Biochemistry at the University of Bologna. She was elected a Fellow of the International Society for Computational Biology (ISCB) in 2020 for outstanding contributions to the fields of computational biology and bioinformatics. Her work in machine learning has been used for protein structure prediction and methods from her group have been highly ranked in international competitions such as the Critical Assessment of protein Structure Prediction (CASP), and the Critical Assessment of Function Annotation (CAFA).
References
- ↑ Haas, J; Roth, S; Arnold, K; Kiefer, F; Schmidt, T; Bordoli, L; Schwede, T (2013). "The Protein Model Portal--a comprehensive resource for protein structure and model information". Database . 2013: bat031. doi:10.1093/database/bat031. PMC 3889916 . PMID 23624946.
- ↑ Moult, J; Fidelis, K; Kryshtafovych, A; Schwede, T; Tramontano, A (February 2014). "Critical assessment of methods of protein structure prediction (CASP)--round x". Proteins . 82 Suppl 2: 1–6. doi:10.1002/prot.24452. PMC 4394854 . PMID 24344053.
- ↑ Mariani, V; Biasini, M; Barbato, A; Schwede, T (1 November 2013). "lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests". Bioinformatics . 29 (21): 2722–8. doi:10.1093/bioinformatics/btt473. PMC 3799472 . PMID 23986568.
- ↑ Arnold, K; Kiefer, F; Kopp, J; Battey, JN; Podvinec, M; Westbrook, JD; Berman, HM; Bordoli, L; Schwede, T (March 2009). "The Protein Model Portal". Journal of Structural and Functional Genomics . 10 (1): 1–8. doi:10.1007/s10969-008-9048-5. PMC 2704613 . PMID 19037750.
- ↑ Gabanyi, MJ; Adams, PD; Arnold, K; Bordoli, L; Carter, LG; Flippen-Andersen, J; Gifford, L; Haas, J; Kouranov, A; McLaughlin, WA; Micallef, DI; Minor, W; Shah, R; Schwede, T; Tao, YP; Westbrook, JD; Zimmerman, M; Berman, HM (July 2011). "The Structural Biology Knowledgebase: a portal to protein structures, sequences, functions, and methods". Journal of Structural and Functional Genomics . 12 (2): 45–54. doi:10.1007/s10969-011-9106-2. PMC 3123456 . PMID 21472436.