
Polymicrogyria (PMG) is a condition that affects the development of the human brain by multiple small gyri (microgyri) creating excessive folding of the brain leading to an abnormally thick cortex. This abnormality can affect either one region of the brain or multiple regions.
Pachygyria is a congenital malformation of the cerebral hemisphere. It results in unusually thick convolutions of the cerebral cortex. Typically, children have developmental delay and seizures, the onset and severity depending on the severity of the cortical malformation. Infantile spasms are common in affected children, as is intractable epilepsy.

Chromosome 15 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 15 spans about 99.7 million base pairs and represents between 3% and 3.5% of the total DNA in cells. Chromosome 15 is an acrocentric chromosome, with a very small short arm, which contains few protein coding genes among its 19 million base pairs. It has a larger long arm that is gene rich, spanning about 83 million base pairs.
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000-100,000 births.
Glucose transporter 1, also known as solute carrier family 2, facilitated glucose transporter member 1 (SLC2A1), is a uniporter protein that in humans is encoded by the SLC2A1 gene. GLUT1 facilitates the transport of glucose across the plasma membranes of mammalian cells. This gene encodes a facilitative glucose transporter that is highly expressed in erythrocytes and endothelial cells, including cells of the blood–brain barrier. The encoded protein is found primarily in the cell membrane and on the cell surface, where it can also function as a receptor for human T-cell leukemia virus (HTLV) I and II. GLUT1 accounts for 2 percent of the protein in the plasma membrane of erythrocytes.
Generalized epilepsy with febrile seizures plus (GEFS+) is a syndromic autosomal dominant disorder where affected individuals can exhibit numerous epilepsy phenotypes. GEFS+ can persist beyond early childhood. GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI), which is also known as Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC). There are at least six types of GEFS+, delineated by their causative gene. Known causative gene mutations are in the sodium channel α subunit genes SCN1A, an associated β subunit SCN1B, and in a GABAA receptor γ subunit gene, in GABRG2 and there is another gene related with calcium channel the PCDH19 which is also known as Epilepsy Female with Mental Retardation. Penetrance for this disorder is estimated at 60%.
Episodic ataxia (EA) is an autosomal dominant disorder characterized by sporadic bouts of ataxia with or without myokymia. There are seven types recognized but the majority are due to two recognized entities. Ataxia can be provoked by psychological stress or startle, or heavy exertion, including exercise. Symptoms can first appear in infancy. There are at least six loci for EA, of which 4 are known genes. Some patients with EA also have migraine or progressive cerebellar degenerative disorders, symptomatic of either familial hemiplegic migraine or spinocerebellar ataxia. Some patients respond to acetazolamide though others do not.

Spinocerebellar ataxia type 13 (SCA13) is a rare autosomal dominant disorder, which, like other types of SCA, is characterized by dysarthria, nystagmus, and ataxia of gait, stance and the limbs due to cerebellar dysfunction. Patients with SCA13 also tend to present with epilepsy, an inability to run, and increased reflexes. This cerebellar dysfunction is permanent and progressive. SCA13 is caused by mutations in KCNC3, a gene encoding a voltage-gated potassium channel KV3.3. There are two known mutations in this gene causative for SCA13. Unlike many other types of SCA, these are not polyglutamine expansions but, rather, point mutations resulting in channels with no current or altered kinetics.

Aristaless related homeobox is a protein that in humans is encoded by the ARX gene.
Dentatorubral–pallidoluysian atrophy (DRPLA) is an autosomal dominant spinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River Syndrome and Naito–Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.

Angelman syndrome or Angelman's syndrome (AS) is a genetic disorder that mainly affects the nervous system. Symptoms include a small head and a specific facial appearance, severe intellectual disability, developmental disability, limited to no functional speech, balance and movement problems, seizures, and sleep problems. Children usually have a happy personality and have a particular interest in water. The symptoms generally become noticeable by one year of age.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
EAST syndrome is a syndrome consisting of epilepsy, ataxia, sensorineural deafness and salt-wasting renal tubulopathy. The tubulopathy in this condition predispose to hypokalemic metabolic alkalosis with normal blood pressure. Hypomagnesemia may also be present.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt–Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.
Cerebral dysgenesis–neuropathy–ichthyosis–keratoderma syndrome is a neurocutaneous condition caused by mutation in the SNAP29 gene.

Renpenning's syndrome is a neurodevelopmental disorder recognised in males that causes intellectual disability, mild growth retardation with examples in the testes and head, and a somewhat short stature. The condition only affects males, starting at birth.
Epilepsy-intellectual disability in females also known as PCDH19 gene-related epilepsy or epileptic encephalopathy, early infantile, 9 (EIEE9), is a rare type of epilepsy that affects predominately females and is characterized by clusters of brief seizures, which start in infancy or early childhood, and is occasionally accompanied by varying degrees of cognitive impairment. The striking pattern of onset seizures at a young age, genetic testing and laboratory results, potential developmental delays or developmental regression and associated disorders, eases diagnosis.
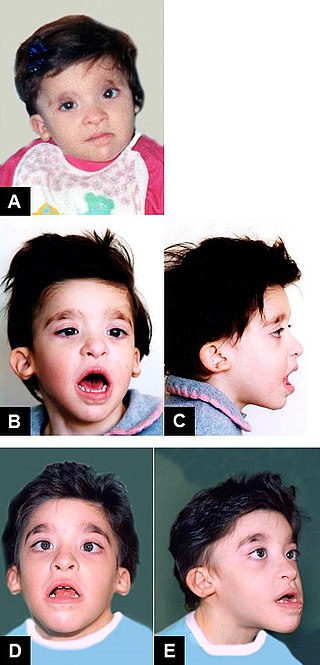
Sanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia, but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere. The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.

Jean-Louis Mandel, born in Strasbourg on February 12, 1946, is a French medical doctor and geneticist, and heads a research team at the Institute of Genetics and Molecular and Cellular Biology (IGBMC). He has been in charge of the genetic diagnosis laboratory at the University Hospitals of Strasbourg since 1992, as well as a professor at the Collège de France since 2003.