Related Research Articles

In the fields of biochemistry and pharmacology an allosteric regulator is a substance that binds to a site on an enzyme or receptor distinct from the active site, resulting in a conformational change that alters the protein's activity, either enhancing or inhibiting its function. In contrast, substances that bind directly to an enzyme's active site or the binding site of the endogenous ligand of a receptor are called orthosteric regulators or modulators.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
Protein design is the rational design of new protein molecules to design novel activity, behavior, or purpose, and to advance basic understanding of protein function. Proteins can be designed from scratch or by making calculated variants of a known protein structure and its sequence. Rational protein design approaches make protein-sequence predictions that will fold to specific structures. These predicted sequences can then be validated experimentally through methods such as peptide synthesis, site-directed mutagenesis, or artificial gene synthesis.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.
Internal Coordinate Mechanics (ICM) is a software program and algorithm to predict low-energy conformations of molecules by sampling the space of internal coordinates defining molecular geometry. In ICM each molecule is constructed as a tree from an entry atom where each next atom is built iteratively from the preceding three atoms via three internal variables. The rings kept rigid or imposed via additional restraints. ICM is used for modelling peptides and interactions with substrates and coenzymes.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.

UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.
Protein–ligand docking is a molecular modelling technique. The goal of protein–ligand docking is to predict the position and orientation of a ligand when it is bound to a protein receptor or enzyme. Pharmaceutical research employs docking techniques for a variety of purposes, most notably in the virtual screening of large databases of available chemicals in order to select likely drug candidates. There has been rapid development in computational ability to determine protein structure with programs such as AlphaFold, and the demand for the corresponding protein-ligand docking predictions is driving implementation of software that can find accurate models. Once the protein folding can be predicted accurately along with how the ligands of various structures will bind to the protein, the ability for drug development to progress at a much faster rate becomes possible.
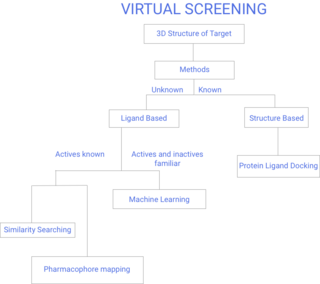
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
In molecular modelling, docking is a method which predicts the preferred orientation of one molecule to another when bound together in a stable complex. In the case of protein docking, the search space consists of all possible orientations of the protein with respect to the ligand. Flexible docking in addition considers all possible conformations of the protein paired with all possible conformations of the ligand.
In the fields of computational chemistry and molecular modelling, scoring functions are mathematical functions used to approximately predict the binding affinity between two molecules after they have been docked. Most commonly one of the molecules is a small organic compound such as a drug and the second is the drug's biological target such as a protein receptor. Scoring functions have also been developed to predict the strength of intermolecular interactions between two proteins or between protein and DNA.
This is a list of notable computer programs that are used for nucleic acids simulations.
Conformational proofreading or conformational selection is a general mechanism of molecular recognition systems, suggested by Yonatan Savir and Tsvi Tlusty, in which introducing an energetic barrier - such as a structural mismatch between a molecular recognizer and its target - enhances the recognition specificity and quality. Conformational proofreading does not require the consumption of energy and may therefore be used in any molecular recognition system. Conformational proofreading is especially useful in scenarios where the recognizer has to select the appropriate target among many similar competitors. Proteins evolve the capacity for conformational proofreading through fine-tuning their geometry, flexibility and chemical interactions with the target.

Ruth Nussinov is an Israeli-American biologist born in Rehovot who works as a professor in the Department of Human Genetics, School of Medicine at Tel Aviv University and is the senior principal scientist and principal investigator at the National Cancer Institute, National Institutes of Health. Nussinov is also the editor in chief of the Current Opinion in Structural Biology and formerly of the journal PLOS Computational Biology.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
LeDock is a molecular docking software, designed for protein-ligand interactions, that is compatible with Linux, macOS, and Windows.

rDock is an open-source molecular docking software that be used for docking small molecules against proteins and nucleic acids. It is primarily designed for high-throughput virtual screening and prediction of binding mode.
References
- ↑ Kuntz, ID; Blaney, JM; Oatley, SJ; Langridge, R; Ferrin, TE (1982). "A geometric approach to macromolecule-ligand interactions". Journal of Molecular Biology. 161 (2): 269–88. doi:10.1016/0022-2836(82)90153-X. PMID 7154081.
- 1 2 Ewing, TJ; Makino, S; Skillman, AG; Kuntz, ID (2001). "DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases". Journal of Computer-aided Molecular Design. 15 (5): 411–28. Bibcode:2001JCAMD..15..411E. doi:10.1023/A:1011115820450. PMID 11394736. S2CID 5553209.
- ↑ Moustakas, DT; Lang, PT; Pegg, S; Pettersen, E; Kuntz, ID; Brooijmans, N; Rizzo, RC (2006). "Development and validation of a modular, extensible docking program: DOCK 5". Journal of Computer-aided Molecular Design. 20 (10–11): 601–19. Bibcode:2006JCAMD..20..601M. doi:10.1007/s10822-006-9060-4. PMID 17149653. S2CID 24495648.
- 1 2 Lang, PT; Brozell, SR; Mukherjee, S; Pettersen, EF; Meng, EC; Thomas, V; Rizzo, RC; Case, DA; et al. (2009). "DOCK 6: Combining techniques to model RNA–small molecule complexes". RNA. 15 (6): 1219–30. doi:10.1261/rna.1563609. PMC 2685511 . PMID 19369428.
- ↑ Lorber, DM; Shoichet, BK (1998). "Flexible ligand docking using conformational ensembles". Protein Sci. 7 (4): 938–950. doi:10.1002/pro.5560070411. PMC 2143983 . PMID 9568900.
- ↑ Lorber, DM; Shoichet, BK (2005). "Hierarchical Docking of Databases of Multiple Ligand Conformations". Curr Top Med Chem. 5 (8): 739–49. doi:10.2174/1568026054637683. PMC 1364474 . PMID 16101414.