
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
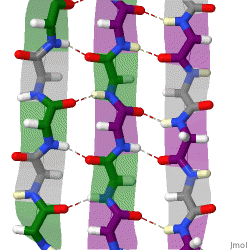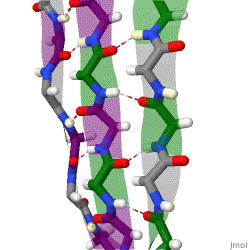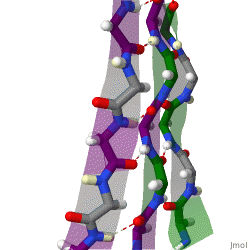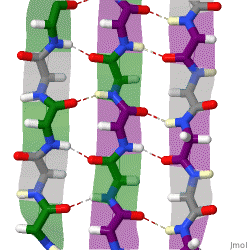
The beta sheet is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

In chemistry, a hydrogen bond is primarily an electrostatic force of attraction between a hydrogen (H) atom which is covalently bonded to a more electronegative "donor" atom or group (Dn), and another electronegative atom bearing a lone pair of electrons—the hydrogen bond acceptor (Ac). Such an interacting system is generally denoted Dn−H···Ac, where the solid line denotes a polar covalent bond, and the dotted or dashed line indicates the hydrogen bond. The most frequent donor and acceptor atoms are the period 2 elements nitrogen (N), oxygen (O), and fluorine (F).
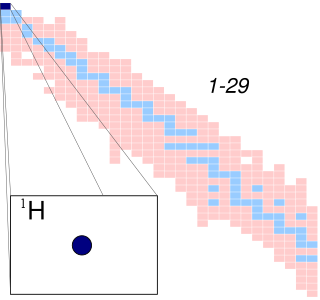
A hydrogen atom is an atom of the chemical element hydrogen. The electrically neutral hydrogen atom contains a nucleus of a single positively charged proton and a single negatively charged electron bound to the nucleus by the Coulomb force. Atomic hydrogen constitutes about 75% of the baryonic mass of the universe.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
Circular dichroism (CD) is dichroism involving circularly polarized light, i.e., the differential absorption of left- and right-handed light. Left-hand circular (LHC) and right-hand circular (RHC) polarized light represent two possible spin angular momentum states for a photon, and so circular dichroism is also referred to as dichroism for spin angular momentum. This phenomenon was discovered by Jean-Baptiste Biot, Augustin Fresnel, and Aimé Cotton in the first half of the 19th century. Circular dichroism and circular birefringence are manifestations of optical activity. It is exhibited in the absorption bands of optically active chiral molecules. CD spectroscopy has a wide range of applications in many different fields. Most notably, UV CD is used to investigate the secondary structure of proteins. UV/Vis CD is used to investigate charge-transfer transitions. Near-infrared CD is used to investigate geometric and electronic structure by probing metal d→d transitions. Vibrational circular dichroism, which uses light from the infrared energy region, is used for structural studies of small organic molecules, and most recently proteins and DNA.
In spectroscopy, the Rydberg constant, symbol for heavy atoms or for hydrogen, named after the Swedish physicist Johannes Rydberg, is a physical constant relating to the electromagnetic spectra of an atom. The constant first arose as an empirical fitting parameter in the Rydberg formula for the hydrogen spectral series, but Niels Bohr later showed that its value could be calculated from more fundamental constants according to his model of the atom.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design.
In polymer science, the polymer chain or simply backbone of a polymer is the main chain of a polymer. Polymers are often classified according to the elements in the main chains. The character of the backbone, i.e. its flexibility, determines the properties of the polymer. For example, in polysiloxanes (silicone), the backbone chain is very flexible, which results in a very low glass transition temperature of −123 °C. The polymers with rigid backbones are prone to crystallization in thin films and in solution. Crystallization in its turn affects the optical properties of the polymers, its optical band gap and electronic levels.
In physics, the Lamb shift, named after Willis Lamb, is an anomalous difference in energy between two electron orbitals in a hydrogen atom. The difference was not predicted by theory and it cannot be derived from the Dirac equation, which predicts identical energies. Hence the Lamb shift is a deviation from theory seen in the differing energies contained by the 2S1/2 and 2P1/2 orbitals of the hydrogen atom.
The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a simple method for calculating molecular orbitals as linear combinations of atomic orbitals. The theory predicts the molecular orbitals for π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine. It provides the theoretical basis for Hückel's rule that cyclic, planar molecules or ions with π-electrons are aromatic. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon and hydrogen (heteroatoms). A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method (EHM), was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general and was used to provide computational justification for the Woodward–Hoffmann rules. To distinguish the original approach from Hoffmann's extension, the Hückel method is also known as the simple Hückel method (SHM).

A protein contact map represents the distance between all possible amino acid residue pairs of a three-dimensional protein structure using a binary two-dimensional matrix. For two residues and , the element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å ; and distance between the side-chain centers of mass.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.

A pi helix is a type of secondary structure found in proteins. Discovered by crystallographer Barbara Low in 1952 and once thought to be rare, short π-helices are found in 15% of known protein structures and are believed to be an evolutionary adaptation derived by the insertion of a single amino acid into an α-helix. Because such insertions are highly destabilizing, the formation of π-helices would tend to be selected against unless it provided some functional advantage to the protein. π-helices therefore are typically found near functional sites of proteins.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.

In quantum biology, the Davydov soliton is a quasiparticle representing an excitation propagating along the self-trapped amide I groups within the α-helices of proteins. It is a solution of the Davydov Hamiltonian.
Helix–coil transition models are formalized techniques in statistical mechanics developed to describe conformations of linear polymers in solution. The models are usually but not exclusively applied to polypeptides as a measure of the relative fraction of the molecule in an alpha helix conformation versus turn or random coil. The main attraction in investigating alpha helix formation is that one encounters many of the features of protein folding but in their simplest version. Most of the helix–coil models contain parameters for the likelihood of helix nucleation from a coil region, and helix propagation along the sequence once nucleated; because polypeptides are directional and have distinct N-terminal and C-terminal ends, propagation parameters may differ in each direction.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of over 15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality, and identifying residue "outliers". The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR's output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.
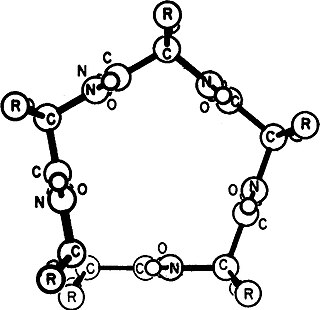
Gamma helix (or γ-helix) is a type of secondary structure in proteins that has been predicted by Pauling, Corey, and Branson, but has never been observed in natural proteins. The hydrogen bond in this type of helix was predicted to be between N-H group of one amino acid and the C=O group of the amino acid six residues earlier (or, as described by Pauling, Corey, Branson, "to the fifth amide group beyond it"). This can also be described as i + 6 → i bond and would be a continuation of the series (310 helix, alpha helix, pi helix and gamma helix). This theoretical helix contains 5.1 residues per turn. However, a fully developed gamma helix has characteristics of a structure that has 2.2 amino acid residues per turn, a rise of 2.75Å per residue, and a pseudo-cyclic (C7) structure closed by intramolecular H-bond. Depending on the amino acid's side chain (R) involved in this main-chain reversal motif, two stereoisomers can occur with their Cα-substituent located either in the axial or in the equatorial position relative to the H-bonded pseudo-cycle.



