In organic chemistry, an acetal is a functional group with the connectivity R2C(OR')2. Here, the R groups can be organic fragments or hydrogen, while the R' groups must be organic fragments not hydrogen. The two R' groups can be equivalent to each other or not. Acetals are formed from and convertible to aldehydes or ketones and have the same oxidation state at the central carbon, but have substantially different chemical stability and reactivity as compared to the analogous carbonyl compounds. The central carbon atom has four bonds to it, and is therefore saturated and has tetrahedral geometry.

Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.
A diol is a chemical compound containing two hydroxyl groups. An aliphatic diol is also called a glycol. This pairing of functional groups is pervasive, and many subcategories have been identified.
The Shapiro reaction or tosylhydrazone decomposition is an organic reaction in which a ketone or aldehyde is converted to an alkene through an intermediate hydrazone in the presence of 2 equivalents of organolithium reagent. The reaction was discovered by Robert H. Shapiro in 1967. The Shapiro reaction was used in the Nicolaou Taxol total synthesis. This reaction is very similar to the Bamford–Stevens reaction, which also involves the basic decomposition of tosyl hydrazones.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.
Silyl ethers are a group of chemical compounds which contain a silicon atom covalently bonded to an alkoxy group. The general structure is R1R2R3Si−O−R4 where R4 is an alkyl group or an aryl group. Silyl ethers are usually used as protecting groups for alcohols in organic synthesis. Since R1R2R3 can be combinations of differing groups which can be varied in order to provide a number of silyl ethers, this group of chemical compounds provides a wide spectrum of selectivity for protecting group chemistry. Common silyl ethers are: trimethylsilyl (TMS), tert-butyldiphenylsilyl (TBDPS), tert-butyldimethylsilyl (TBS/TBDMS) and triisopropylsilyl (TIPS). They are particularly useful because they can be installed and removed very selectively under mild conditions.

The Wieland–Miescher ketone is a racemic bicyclic diketone (enedione) and is a versatile synthon which has so far been employed in the total synthesis of more than 50 natural products, predominantly sesquiterpenoids, diterpenes and steroids possessing possible biological properties including anticancer, antimicrobial, antiviral, antineurodegenerative and immunomodulatory activities. The reagent is named after two chemists from Ciba Geigy, Karl Miescher and Peter Wieland. Examples of syntheses performed using the optically active enantiomer of this diketone as a starting material are that of ancistrofuran and the Danishefsky total synthesis of Taxol.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.

The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994, was the first total synthesis of Taxol.
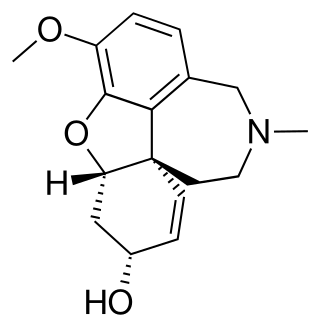
The article concerns the total synthesis of galanthamine, a drug used for the treatment of mild to moderate Alzheimer's disease.

Epothilones are a class of potential cancer drugs. Like taxanes, they prevent cancer cells from dividing by interfering with tubulin, but in early trials, epothilones have better efficacy and milder adverse effects than taxanes.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
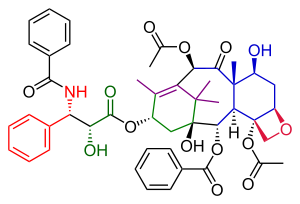
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

The Kuwajima Taxol total synthesis by the group of Isao Kuwajima of the Tokyo Institute of Technology is one of several efforts in taxol total synthesis published in the 1990s. The total synthesis of Taxol is considered a landmark in organic synthesis.

The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.
The Parikh–Doering oxidation is an oxidation reaction that transforms primary and secondary alcohols into aldehydes and ketones, respectively. The procedure uses dimethyl sulfoxide (DMSO) as the oxidant and the solvent, activated by the sulfur trioxide pyridine complex (SO3•C5H5N) in the presence of triethylamine or diisopropylethylamine as base. Dichloromethane is frequently used as a cosolvent for the reaction.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
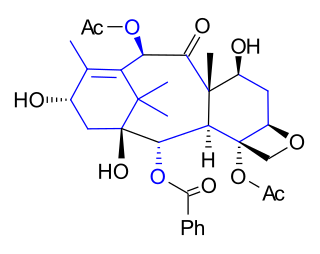
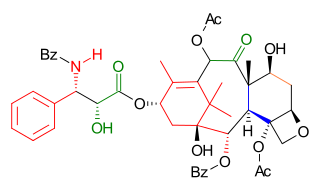
The Takahashi Taxol total synthesis published by Takashi Takahashi in 2006 is one of several methods in taxol total synthesis. The method starts from geraniol and differs from the other 6 published methods that it is a formal synthesis and that it is racemic. A key feature of the published procedure is that several synthetic steps were performed in an automated synthesizer on a scale up to 300 gram and that purification steps were also automated.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.

Teruaki Mukaiyama was a Japanese organic chemist. One of the most prolific chemists of the 20th century in the field of organic synthesis, Mukaiyama helped establish the field of organic chemistry in Japan after World War II.