Related Research Articles
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations. The PDB is overseen by an organization called the Worldwide Protein Data Bank, wwPDB.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
BioJava is an open-source software project dedicated to provide Java tools to process biological data. BioJava is a set of library functions written in the programming language Java for manipulating sequences, protein structures, file parsers, Common Object Request Broker Architecture (CORBA) interoperability, Distributed Annotation System (DAS), access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a Protein Data Bank (PDB) file, interacting with Jmol and many more. This application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.

Amos Bairoch is a Swiss bioinformatician and Professor of Bioinformatics at the Department of Human Protein Sciences of the University of Geneva where he leads the CALIPHO group at the Swiss Institute of Bioinformatics (SIB) combining bioinformatics, curation, and experimental efforts to functionally characterize human proteins.
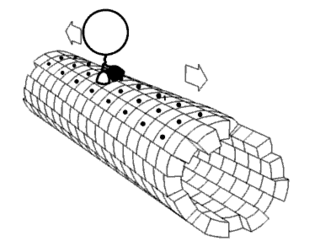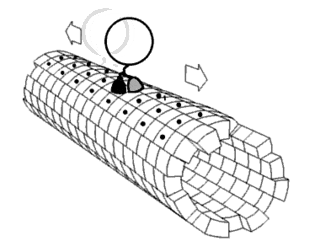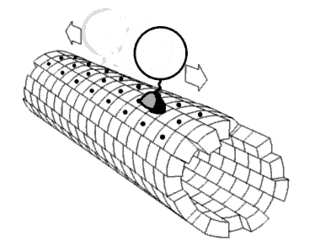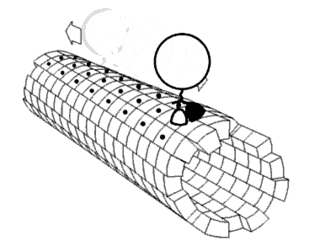
In biochemistry, a conformational change is a change in the shape of a macromolecule, often induced by environmental factors.

PROSITE is a protein database. It consists of entries describing the protein families, domains and functional sites as well as amino acid patterns and profiles in them. These are manually curated by a team of the Swiss Institute of Bioinformatics and tightly integrated into Swiss-Prot protein annotation. PROSITE was created in 1988 by Amos Bairoch, who directed the group for more than 20 years. Since July 2018, the director of PROSITE and Swiss-Prot is Alan Bridge.
Expasy is an online bioinformatics resource operated by the SIB Swiss Institute of Bioinformatics. It is an extensible and integrative portal which provides access to over 160 databases and software tools and supports a range of life science and clinical research areas, from genomics, proteomics and structural biology, to evolution and phylogeny, systems biology and medical chemistry. The individual resources are hosted in a decentralized way by different groups of the SIB Swiss Institute of Bioinformatics and partner institutions.
Mark Bender Gerstein is an American scientist working in bioinformatics and Data Science. As of 2009, he is co-director of the Yale Computational Biology and Bioinformatics program.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.
SWISS-MODEL is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. Homology modeling is currently the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures ("templates") to build models for evolutionary related proteins ("targets").

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.
Werner G. Krebs is an American data scientist. He is currently CEO of data science and artificial intelligence startup Acculation, Inc. and has previously held positions at what are now Virtu Financial, Bank of America, and the San Diego Supercomputer Center.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.
References
- 1 2 Gerstein M, Krebs W (September 1998). "A database of macromolecular motions". Nucleic Acids Res. 26 (18): 4280–90. doi:10.1093/nar/26.18.4280. PMC 147832 . PMID 9722650.
- 1 2 Flores, S; Echols, N; Milburn, D; Hespenheide, B; Keating, K; Lu, J; Wells, S; Yu, E. Z.; Thorpe, M; Gerstein, M (2006). "The Database of Macromolecular Motions: new features added at the decade mark". Nucleic Acids Research. 34 (Database issue): D296–301. doi:10.1093/nar/gkj046. PMC 1347409 . PMID 16381870.
- 1 2 Krebs WG, Gerstein M (April 2000). "SURVEY AND SUMMARY: The morph server: a standardized system for analyzing and visualizing macromolecular motions in a database framework". Nucleic Acids Res. 28 (8): 1665–75. doi:10.1093/nar/28.8.1665. PMC 102811 . PMID 10734184.
- 1 2 "Hot Picks". Science. 284 (5416): 871b–871. 1999-05-07. doi:10.1126/science.284.5416.871b. ISSN 0036-8075. S2CID 220104660.
- 1 2 Bourne PE, Helge W, eds. (2003). Structural Bioinformatics . Hoboken, NJ: Wiley-Liss. p. 229. ISBN 978-0-471-20199-1. OCLC 50199108.
- 1 2 Bourne, PE; Murray-Rust, J; Lakey JH (Feb 1999). "Protein-nucleic acid interactions Folding and binding Web alert". Current Opinion in Structural Biology. 9 (1): 9–10. doi:10.1016/S0959-440X(99)90000-3.
- 1 2 "Morphs". Proteopeida. Retrieved 2015-10-30.
- 1 2 Borner (ed.). Knowledge Management and Visualization Tools in Support of Discovery (NSF Workshop Report) (PDF) (Report). National Science Foundation Workshop. p. 5. Archived from the original (PDF) on 2016-03-04. Retrieved 2015-12-26.
- ↑ "Mark Gerstein - Google Scholar Citations". scholar.google.com. Retrieved 2015-12-26.
- ↑ "Werner G. Krebs - Google Scholar Citations". scholar.google.com. Retrieved 2015-12-26.
- 1 2 Maiti R, Van Domselaar GH, Wishart DS (July 2005). "MovieMaker: a web server for rapid rendering of protein motions and interactions". Nucleic Acids Res. 33 (Web Server issue): W358–62. doi:10.1093/nar/gki485. PMC 1160245 . PMID 15980488.
- ↑ Alexandrov V, Gerstein M (January 2004). "Using 3D Hidden Markov Models that explicitly represent spatial coordinates to model and compare protein structures". BMC Bioinformatics. 5: 2. doi: 10.1186/1471-2105-5-2 . PMC 344530 . PMID 14715091.
- ↑ Alexandrov V, Lehnert U, Echols N, Milburn D, Engelman D, Gerstein M (March 2005). "Normal modes for predicting protein motions: A comprehensive database assessment and associated Web tool". Protein Sci. 14 (3): 633–43. doi:10.1110/ps.04882105. PMC 2279292 . PMID 15722444.
- ↑ "The Motion Library in Protean3D documentation". www.dnastar.com. Retrieved 2015-11-13.
- ↑ "Protean3D overview". www.dnastar.com. Retrieved 2015-11-13.