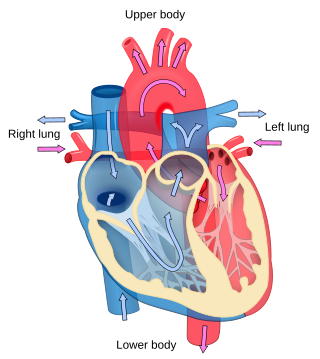
Cardiology is the study of the heart. Cardiology is a branch of medicine that deals with disorders of the heart and the cardiovascular system. The field includes medical diagnosis and treatment of congenital heart defects, coronary artery disease, heart failure, valvular heart disease, and electrophysiology. Physicians who specialize in this field of medicine are called cardiologists, a sub-specialty of internal medicine. Pediatric cardiologists are pediatricians who specialize in cardiology. Physicians who specialize in cardiac surgery are called cardiothoracic surgeons or cardiac surgeons, a specialty of general surgery.

Heart murmurs are unique heart sounds produced when blood flows across a heart valve or blood vessel. This occurs when turbulent blood flow creates a sound loud enough to hear with a stethoscope. The sound differs from normal heart sounds by their characteristics. For example, heart murmurs may have a distinct pitch, duration and timing. The major way health care providers examine the heart on physical exam is heart auscultation; another clinical technique is palpation, which can detect by touch when such turbulence causes the vibrations called cardiac thrill. A murmur is a sign found during the cardiac exam. Murmurs are of various types and are important in the detection of cardiac and valvular pathologies.

Tetralogy of Fallot (TOF), formerly known as Steno-Fallot tetralogy, is a congenital heart defect characterized by four specific cardiac defects. Classically, the four defects are:

Patent ductus arteriosus (PDA) is a medical condition in which the ductus arteriosus fails to close after birth: this allows a portion of oxygenated blood from the left heart to flow back to the lungs through the aorta, which has a higher blood pressure, to the pulmonary artery, which has a lower blood pressure. Symptoms are uncommon at birth and shortly thereafter, but later in the first year of life there is often the onset of an increased work of breathing and failure to gain weight at a normal rate. With time, an uncorrected PDA usually leads to pulmonary hypertension followed by right-sided heart failure.

Atrial septal defect (ASD) is a congenital heart defect in which blood flows between the atria of the heart. Some flow is a normal condition both pre-birth and immediately post-birth via the foramen ovale; however, when this does not naturally close after birth it is referred to as a patent (open) foramen ovale (PFO). It is common in patients with a congenital atrial septal aneurysm (ASA).

Pulmonary hypertension is a condition of increased blood pressure in the arteries of the lungs. Symptoms include shortness of breath, fainting, tiredness, chest pain, swelling of the legs, and a fast heartbeat. The condition may make it difficult to exercise. Onset is typically gradual. According to the definition at the 6th World Symposium of Pulmonary Hypertension in 2018, a patient is deemed to have pulmonary hypertension if the pulmonary mean arterial pressure is greater than 20mmHg at rest, revised down from a purely arbitrary 25mmHg, and pulmonary vascular resistance (PVR) greater than 3 Wood units.

A congenital heart defect (CHD), also known as a congenital heart anomaly, congenital cardiovascular malformation, and congenital heart disease, is a defect in the structure of the heart or great vessels that is present at birth. A congenital heart defect is classed as a cardiovascular disease. Signs and symptoms depend on the specific type of defect. Symptoms can vary from none to life-threatening. When present, symptoms are variable and may include rapid breathing, bluish skin (cyanosis), poor weight gain, and feeling tired. CHD does not cause chest pain. Most congenital heart defects are not associated with other diseases. A complication of CHD is heart failure.

A ventricular septal defect (VSD) is a defect in the ventricular septum, the wall dividing the left and right ventricles of the heart. The extent of the opening may vary from pin size to complete absence of the ventricular septum, creating one common ventricle. The ventricular septum consists of an inferior muscular and superior membranous portion and is extensively innervated with conducting cardiomyocytes.
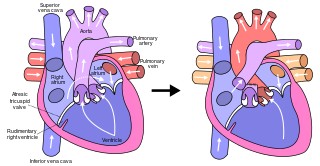
The Fontan procedure or Fontan–Kreutzer procedure is a palliative surgical procedure used in children with univentricular hearts. It involves diverting the venous blood from the inferior vena cava (IVC) and superior vena cava (SVC) to the pulmonary arteries. The procedure varies for differing congenital heart pathologies. For example, in tricuspid atresia, the procedure can be done where the blood does not pass through the morphologic right ventricle; i.e., the systemic and pulmonary circulations are placed in series with the functional single ventricle. By contrast, in hypoplastic left heart syndrome, the heart is more reliant on the more functional right ventricle to provide blood flow to the systemic circulation. The procedure was initially performed in 1968 by Francis Fontan and Eugene Baudet from Bordeaux, France, published in 1971, simultaneously described in July 1971 by Guillermo Kreutzer from Buenos Aires, Argentina, presented at the Argentinean National Cardilogy meeting of that year and finally published in 1973.
Situs ambiguus, or heterotaxy, is a rare congenital defect in which the major visceral organs are distributed abnormally within the chest and abdomen. Clinically, heterotaxy spectrum generally refers to any defect of left-right asymmetry and arrangement of the visceral organs; however, classical heterotaxy requires multiple organs to be affected. This does not include the congenital defect situs inversus, which results when arrangement of all the organs in the abdomen and chest are mirrored, so the positions are opposite the normal placement. Situs inversus is the mirror image of situs solitus, which is normal asymmetric distribution of the abdominothoracic visceral organs. Situs ambiguus can also be subdivided into left-isomerism and right isomerism based on the defects observed in the spleen, lungs and atria of the heart.
The Rastelli procedure is an open heart surgical procedure developed by Italian physician and cardiac surgery researcher, Giancarlo Rastelli, in 1967 at the Mayo Clinic, and involves using a pulmonary or aortic homograft conduit to relieve pulmonary obstruction in double outlet right ventricle with pulmonary stenosis.
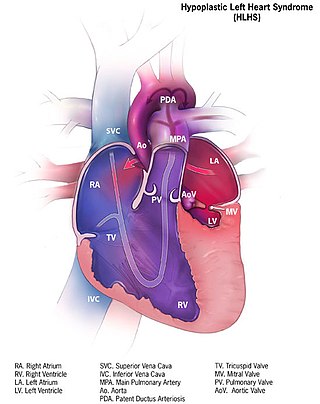
Hypoplastic left heart syndrome (HLHS) is a rare congenital heart defect in which the left side of the heart is severely underdeveloped and incapable of supporting the systemic circulation. It is estimated to account for 2-3% of all congenital heart disease. Early signs and symptoms include poor feeding, cyanosis, and diminished pulse in the extremities. The etiology is believed to be multifactorial resulting from a combination of genetic mutations and defects resulting in altered blood flow in the heart. Several structures can be affected including the left ventricle, aorta, aortic valve, or mitral valve all resulting in decreased systemic blood flow.

Pulmonary atresia is a congenital malformation of the pulmonary valve in which the valve orifice fails to develop. The valve is completely closed thereby obstructing the outflow of blood from the heart to the lungs. The pulmonary valve is located on the right side of the heart between the right ventricle and pulmonary artery. In a normal functioning heart, the opening to the pulmonary valve has three flaps that open and close.

Atrioventricular septal defect (AVSD) or atrioventricular canal defect (AVCD), also known as "common atrioventricular canal" or "endocardial cushion defect" (ECD), is characterized by a deficiency of the atrioventricular septum of the heart that creates connections between all four of its chambers. It is a very specific combination of 3 defects:
A right-to-left shunt is a cardiac shunt which allows blood to flow from the right heart to the left heart. This terminology is used both for the abnormal state in humans and for normal physiological shunts in reptiles.

Right ventricular hypertrophy (RVH) is a condition defined by an abnormal enlargement of the cardiac muscle surrounding the right ventricle. The right ventricle is one of the four chambers of the heart. It is located towards the right lower chamber of the heart and it receives Deoxygenated blood from the right upper chamber and pumps blood into the lungs.
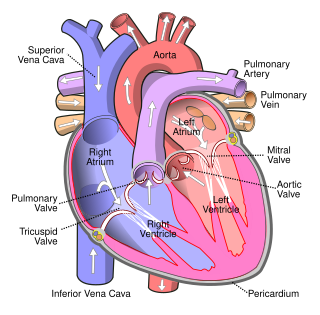
Lutembacher's syndrome is a very rare form of congenital heart disease that affects one of the chambers of the heart as well as a valve. It is commonly known as both congenital atrial septal defect (ASD) and acquired mitral stenosis (MS). Congenital atrial septal defect refers to a hole being in the septum or wall that separates the two atria; this condition is usually seen in fetuses and infants. Mitral stenosis refers to mitral valve leaflets sticking to each other making the opening for blood to pass from the atrium to the ventricles very small. With the valve being so small, blood has difficulty passing from the left atrium into the left ventricle. Septal defects that may occur with Lutembacher's syndrome include: Ostium primum atrial septal defect or ostium secundum which is more prevalent.

Aortopulmonary window (APW) is a faulty connection between the aorta and the main pulmonary artery that results in a significant left-to-right shunt. The aortopulmonary window is the rarest of septal defects, accounting for 0.15-0.6% of all congenital heart malformations. An aortopulmonary window can develop alone or in up to 50% of cases alongside other cardiac defects such as interrupted aortic arch, coarctation of the aorta, transposition of great vessels, and tetralogy of Fallot.

Pulmonary atresia with ventricular septal defect is a rare birth defect characterized by pulmonary valve atresia occurring alongside a defect on the right ventricular outflow tract.
Raghib syndrome is rare a congenital heart defect where the left superior vena cava (LSVC) is draining into the left atrium in addition to an absent coronary sinus and an atrial septal defect. This can be considered a dangerous heart condition because it puts the individual at a high risk of stroke. Other defects that are often associated with Raghib syndrome can include ventricular septal defects, enlargement of the tricuspid annulus, and pulmonary stenosis. While this is considered an extremely rare developmental complex, cases regarding a persistent left superior vena cava (PLSVC) are relatively common among congenital heart defects. It is also important to note that the PLSVC often drains into the right atrium, and only drains into the left atrium in approximately 10 to 20% of individuals with the defect.

