Related Research Articles

Epilepsy is a group of non-communicable neurological disorders characterized by recurrent epileptic seizures. An epileptic seizure is the clinical manifestation of an abnormal, excessive, purposeless and synchronized electrical discharge in the brain cells called neurons. The occurrence of two or more unprovoked seizures defines epilepsy. The occurrence of just one seizure may warrant the definition in a more clinical usage where recurrence may be able to be prejudged. Epileptic seizures can vary from brief and nearly undetectable periods to long periods of vigorous shaking due to abnormal electrical activity in the brain. These episodes can result in physical injuries, either directly such as broken bones or through causing accidents. In epilepsy, seizures tend to recur and may have no immediate underlying cause. Isolated seizures that are provoked by a specific cause such as poisoning are not deemed to represent epilepsy. People with epilepsy may be treated differently in various areas of the world and experience varying degrees of social stigma due to the alarming nature of their symptoms.
An epileptic seizure, informally known as a seizure, is a period of symptoms due to abnormally excessive or synchronous neuronal activity in the brain. Outward effects vary from uncontrolled shaking movements involving much of the body with loss of consciousness, to shaking movements involving only part of the body with variable levels of consciousness, to a subtle momentary loss of awareness. Most of the time these episodes last less than two minutes and it takes some time to return to normal. Loss of bladder control may occur.

A febrile seizure, also known as a fever fit or febrile convulsion, is a seizure associated with a high body temperature but without any serious underlying health issue. They most commonly occur in children between the ages of 6 months and 5 years. Most seizures are less than five minutes in duration, and the child is completely back to normal within an hour of the event. There are two types: simple febrile seizures and complex febrile seizures. Simple febrile seizures involve an otherwise healthy child who has at most one tonic-clonic seizure lasting less than 15 minutes in a 24-hour period. Complex febrile seizures have focal symptoms, last longer than 15 minutes, or occur more than once within 24 hours. About 80% are classified as simple febrile seizures.
Status epilepticus (SE), or status seizure, is a medical condition consisting of a single seizure lasting more than 5 minutes, or 2 or more seizures within a 5-minute period without the person returning to normal between them. Previous definitions used a 30-minute time limit. The seizures can be of the tonic–clonic type, with a regular pattern of contraction and extension of the arms and legs, or of types that do not involve contractions, such as absence seizures or complex partial seizures. Status epilepticus is a life-threatening medical emergency, particularly if treatment is delayed.
Lennox–Gastaut syndrome (LGS) is a complex, rare, and severe childhood-onset epilepsy syndrome. It is characterized by multiple and concurrent seizure types including tonic seizure, cognitive dysfunction, and slow spike waves on electroencephalogram (EEG), which are very abnormal. Typically, it presents in children aged 3–5 years and most of the time persists into adulthood with slight changes in the electroclinical phenotype. It has been associated with perinatal injuries, congenital infections, brain malformations, brain tumors, genetic disorders such as tuberous sclerosis and numerous gene mutations. Sometimes LGS is observed after infantile epileptic spasm syndrome. The prognosis for LGS is marked by a 5% mortality in childhood and persistent seizures into adulthood.

In the field of neurology, temporal lobe epilepsy is an enduring brain disorder that causes unprovoked seizures from the temporal lobe. Temporal lobe epilepsy is the most common type of focal onset epilepsy among adults. Seizure symptoms and behavior distinguish seizures arising from the medial temporal lobe from seizures arising from the lateral (neocortical) temporal lobe. Memory and psychiatric comorbidities may occur. Diagnosis relies on electroencephalographic (EEG) and neuroimaging studies. Anticonvulsant medications, epilepsy surgery and dietary treatments may improve seizure control.
Sudden unexpected death in epilepsy (SUDEP) is a fatal complication of epilepsy. It is defined as the sudden and unexpected, non-traumatic and non-drowning death of a person with epilepsy, without a toxicological or anatomical cause of death detected during the post-mortem examination.
In the field of neurology, seizure types are categories of seizures defined by seizure behavior, symptoms, and diagnostic tests. The International League Against Epilepsy (ILAE) 2017 classification of seizures is the internationally recognized standard for identifying seizure types. The ILAE 2017 classification of seizures is a revision of the prior ILAE 1981 classification of seizures. Distinguishing between seizure types is important since different types of seizures may have different causes, outcomes, and treatments.

Sultiame is a sulfonamide and inhibitor of the enzyme carbonic anhydrase. It is used as an anticonvulsant.
Dravet syndrome (DS), previously known as severe myoclonic epilepsy of infancy (SMEI), is an autosomal dominant genetic disorder which causes a catastrophic form of epilepsy, with prolonged seizures that are often triggered by hot temperatures or fever. It is very difficult to treat with anticonvulsant medications. It often begins before one year of age, with six months being the age that seizures, characterized by prolonged convulsions and triggered by fever, usually begin.
Childhood absence epilepsy (CAE), formerly known as pyknolepsy, is an idiopathic generalized epilepsy which occurs in otherwise normal children. The age of onset is between 4–10 years with peak age between 5–7 years. Children have absence seizures which although brief, they occur frequently, sometimes in the hundreds per day. The absence seizures of CAE involve abrupt and severe impairment of consciousness. Mild automatisms are frequent, but major motor involvement early in the course excludes this diagnosis. The EEG demonstrates characteristic "typical 3Hz spike-wave" discharges. The presence of any other seizure type at time of diagnosis rules out the diagnose of CAE. Prognosis is usually good in well-defined cases of CAE with most patients "growing out" of their epilepsy.
Ohtahara syndrome (OS), also known as early infantile epileptic encephalopathy (EIEE) is a progressive epileptic encephalopathy. The syndrome is outwardly characterized by tonic spasms and partial seizures within the first few months of life, and receives its more elaborate name from the pattern of burst activity on an electroencephalogram (EEG). It is an extremely debilitating progressive neurological disorder, involving intractable seizures and severe intellectual disabilities. No single cause has been identified, although in many cases structural brain damage is present.
Spike-and-wave is a pattern of the electroencephalogram (EEG) typically observed during epileptic seizures. A spike-and-wave discharge is a regular, symmetrical, generalized EEG pattern seen particularly during absence epilepsy, also known as ‘petit mal’ epilepsy. The basic mechanisms underlying these patterns are complex and involve part of the cerebral cortex, the thalamocortical network, and intrinsic neuronal mechanisms.
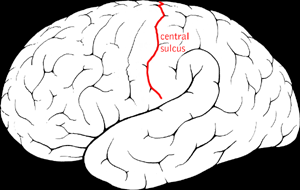
Benign Rolandic epilepsy or self-limited epilepsy with centrotemporal spikes is the most common epilepsy syndrome in childhood. Most children will outgrow the syndrome, hence the label benign. The seizures, sometimes referred to as sylvian seizures, start around the central sulcus of the brain.
Epilepsy-intellectual disability in females also known as PCDH19 gene-related epilepsy or epileptic encephalopathy, early infantile, 9 (EIEE9), is a rare type of epilepsy that affects predominately females and is characterized by clusters of brief seizures, which start in infancy or early childhood, and is occasionally accompanied by varying degrees of cognitive impairment. The striking pattern of onset seizures at a young age, genetic testing and laboratory results, potential developmental delays or developmental regression and associated disorders, eases diagnosis.
Early myoclonic encephalopathy (EME) is a rare neonatal-onset epilepsy developmental and epileptic encephalopathy (DEE) with an onset at neonatal period or during the first 3 months of life. This syndrome is now included as part of the Early infantile developmental and epileptic encephalopathy (EIDEE) under the 2022 ILAE syndrome classification.
People with epilepsy may be classified into different syndromes based on specific clinical features. These features include the age at which seizures begin, the seizure types, and EEG findings, among others. Identifying an epilepsy syndrome is useful as it helps determine the underlying causes as well as deciding what anti-seizure medication should be tried. Epilepsy syndromes are more commonly diagnosed in infants and children. Some examples of epilepsy syndromes include benign rolandic epilepsy, childhood absence epilepsy and juvenile myoclonic epilepsy. Severe syndromes with diffuse brain dysfunction caused, at least partly, by some aspect of epilepsy, are also referred to as epileptic encephalopathies. These are associated with frequent seizures that are resistant to treatment and severe cognitive dysfunction, for instance Lennox-Gastaut syndrome and West syndrome.
A neonatal seizure is a seizure in a baby younger than age 4-weeks that is identifiable by an electrical recording of the brain. It is an occurrence of abnormal, paroxysmal, and persistent ictal rhythm with an amplitude of 2 microvolts in the electroencephalogram,. These may be manifested in form of stiffening or jerking of limbs or trunk. Sometimes random eye movements, cycling movements of legs, tonic eyeball movements, and lip-smacking movements may be observed. Alteration in heart rate, blood pressure, respiration, salivation, pupillary dilation, and other associated paroxysmal changes in the autonomic nervous system of infants may be caused due to these seizures. Often these changes are observed along with the observance of other clinical symptoms. A neonatal seizure may or may not be epileptic. Some of them may be provoked. Most neonatal seizures are due to secondary causes. With hypoxic ischemic encephalopathy being the most common cause in full term infants and intraventricular hemorrhage as the most common cause in preterm infants.
Drug-resistant epilepsy (DRE), also known as refractory epilepsy, intractable epilepsy, or pharmacoresistant epilepsy, is diagnosed following a failure of adequate trials of two tolerated and appropriately chosen and used antiepileptic drugs (AEDs) to achieve sustained seizure freedom. The probability that the next medication will achieve seizure freedom drops with every failed AED. For example, after two failed AEDs, the probability that the third will achieve seizure freedom is around 4%. Drug-resistant epilepsy is commonly diagnosed after several years of uncontrolled seizures, however, in most cases, it is evident much earlier. Approximately 30% of people with epilepsy have a drug-resistant form.
Malignant migrating partial seizures of infancy (MMPSI) is a rare epileptic syndrome that onsets before 6 months of age, commonly in the first few weeks of life. Once seizures start, the site of seizure activity repeatedly migrates from one area of the brain to another, with few periods of remission in between. These seizures are 'focal' (updated term for 'partial'), meaning they do not affect both sides of the brain at the same time. These continuous seizures cause damage to the brain, hence the descriptor 'malignant.'
References
- 1 2 3 4 5 6 7 8 9 10 11 12 "Febrile infection related epilepsy". www.epilepsydiagnosis.org. ILAE. Archived from the original on 2 August 2023. Retrieved 9 September 2023.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Nabbout, Rima. "Orphanet: Febrile infection related epilepsy syndrome". www.orpha.net. Archived from the original on 31 March 2023. Retrieved 9 September 2023.
- ↑ "Febrile infection related epilepsy". www.epilepsydiagnosis.org. Archived from the original on 11 August 2022. Retrieved 9 September 2023.
- ↑ "FEBRILE INFECTION RELATED EPILEPSY". www.epilepsydiagnosis.org. Archived from the original on 11 August 2022. Retrieved 9 September 2023.
- 1 2 3 4 Specchio, Nicola; Pietrafusa, Nicola (August 2020). "New‐onset refractory status epilepticus and febrile infection‐related epilepsy syndrome". Developmental Medicine & Child Neurology. 62 (8): 897–905. doi: 10.1111/dmcn.14553 . PMID 32372459. S2CID 218520258.
- 1 2 3 Kramer, U; Chi, CS; Lin, KL; Specchio, N; Sahin, M; Olson, H; Nabbout, R; Kluger, G; Lin, JJ; van Baalen, A (November 2011). "Febrile infection-related epilepsy syndrome (FIRES): pathogenesis, treatment, and outcome: a multicenter study on 77 children". Epilepsia. 52 (11): 1956–65. doi: 10.1111/j.1528-1167.2011.03250.x . PMID 21883180.
- ↑ Hirsch, LU; Gaspard, N; van Baalen, A; Nabbout, R; Demeret, S; Loddenkemper, T; Navarro, N; Specchio, N; Lagae, L; Rossetti, A; Hocker, S; Gofton, TE; Abend, NS; Gilmore, EJ; Hahn, C; Khosravani, H; Rosenow, F; Trinka, E (April 2018). "Proposed consensus definitions for new‐onset refractory status epilepticus (NORSE), febrile infection‐related epilepsy syndrome (FIRES), and related conditions". Epilepsia. 59 (4): 739–744. doi: 10.1111/epi.14016 . PMID 29399791. S2CID 4495659.
- ↑ "FEBRILE INFECTION RELATED EPILEPSY". www.epilepsydiagnosis.org. Archived from the original on 12 October 2014. Retrieved 7 October 2014.
- ↑ Appenzeller, S; Helbig, I; Stephani, U; Haeusler, M; Kluger, G; Bungeroth, M; Müller, S; Kuhlenbäumer, G; Van Baalen, A (2012). "Febrile infection-related epilepsy syndrome (FIRES) is not caused by SCN1A, POLG, PCDH19 mutations or rare copy number variations". Developmental Medicine & Child Neurology. 54 (12): 1144–1148. doi: 10.1111/j.1469-8749.2012.04435.x . PMID 23066759. S2CID 19954159.
- ↑ van Baalen, A; Häusler, M; Boor, R; Rohr, A; Sperner, J; Kurlemann, G; Panzer, A; Stephani, U; Kluger, G (2010). "Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood". Epilepsia. 51 (7): 1323–1328. doi: 10.1111/j.1528-1167.2010.02535.x . PMID 20345937. S2CID 27421912.
- ↑ Fox, Kristy; Wells, Mary Ellen; Tennison, Michael; Vaughn, Bradley (July 11, 2017). "Febrile Infection-Related Epilepsy Syndrome (FIRES): A Literature Review and Case Study". The Neurodiagnostic Journal. 57 (3): 224–233. doi:10.1080/21646821.2017.1355181. PMID 28898171. S2CID 8944135. Archived from the original on August 9, 2021. Retrieved August 18, 2023.
- ↑ Howell, KB; Katanyuwong, K; Mackay, MT; Bailey, CA; Scheffer, IE; Freeman, JL; Berkovic, SF; Harvey, A (2012). "Long-term follow-up of febrile infection-related epilepsy syndrome". Epilepsia. 53 (1): 101–110. doi: 10.1111/j.1528-1167.2011.03350.x . PMID 22191582. S2CID 9165846.
- ↑ Nabbout, R; Vezzani, A; Dulac, O; Chiron, C (2011). "Acute encephalopathy with inflammation-mediated status epilepticus". Lancet Neurol. 10 (1): 99–108. doi:10.1016/S1474-4422(10)70214-3. PMID 21163447. S2CID 206159592.
- ↑ Carabello, RH; Reyes, G; Avaria, MF; Buompadre, MC; Gonzalez, M; Fortini, S; Cersosimo, R (2013). "Febrile infection related epilepsy syndrome: a study of 12 patients". Seizure. 22 (7): 553–559. doi: 10.1016/j.seizure.2013.04.005 . PMID 23643626. S2CID 14416943.
- ↑ Pranzatelli, MR; Nadi, NS (1995). "Mechanism of action of antiepileptic and antimyoclonic drugs". Advances in Neurology. 67: 329–60. PMID 8848979.
- ↑ Mikaeloff, Y; Jambaqué, I; Hertz-Pannier, L; Zamfirescu, A; Adamsbaum, C; Pluin, P; Dulac, O; Chiron, C (2006). "Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis". Epilepsy Res. 69 (1): 67–79. doi:10.1016/j.eplepsyres.2006.01.002. PMID 16469483. S2CID 25351321.
- ↑ Nabbout, R; Mazzuca, M; Hubert, P; Peudennier, S; Allaire, C; Flurin, V; Aberastury, M; Silva, W; Dulac, O (2010). "Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES)". Epilepsia. 51 (10): 2033–2037. doi:10.1111/j.1528-1167.2010.02703.x. PMID 20813015. S2CID 19453163.
- ↑ van Baalen, A; Häusler, M; Boor, R; Rohr, A; Sperner, J; Kurlemann, G; Panzer, A; Stephani, U; Kluger, G (July 2010). "Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood". Epilepsia. 51 (7): 1323–8. doi: 10.1111/j.1528-1167.2010.02535.x . PMID 20345937. S2CID 27421912.
- 1 2 Simon Shorvon and Monica Ferlisi (2011-09-13). "The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol". Brain. Brain.oxfordjournals.org. 134 (Pt 10): 2802–2818. doi: 10.1093/brain/awr215 . PMID 21914716.