
A rhabdomyoma is a benign tumor of striated muscle. Rhabdomyomas may be either cardiac or extracardiac. Extracardiac forms of rhabdomyoma are sub-classified into three distinct types: adult type, fetal type, and genital type.

Hemangioendotheliomas are a family of vascular neoplasms of intermediate malignancy.

Canalicular adenoma is a benign, epithelial salivary gland neoplasm arranged in interconnecting cords of columnar cells. This is a very rare benign neoplasm, that makes up about 1% of all salivary gland tumors, or about 4% of all benign salivary gland tumors.

A schwannoma is a usually benign nerve sheath tumor composed of Schwann cells, which normally produce the insulating myelin sheath covering peripheral nerves.

Granular cell tumor is a tumor that can develop on any skin or mucosal surface, but occurs on the tongue 40% of the time.
Alveolar rhabdomyosarcoma (ARMS) is a subtype of the rhabdomyosarcoma soft tissue cancer family whose lineage is from mesenchymal cells and are related to skeletal muscle cells. ARMS tumors resemble the alveolar tissue in the lungs. Tumor location varies from patient to patient, but is commonly found in the head and neck region, male and female urogenital tracts, the torso, and extremities. Two fusion proteins can be associated with ARMS, but are not necessary, PAX3-FKHR. and PAX7-FKHR. In children and adolescents ARMS accounts for about 1 percent of all malignancies, has an incidence rate of 1 per million, and most cases occur sporadically with no genetic predisposition. PAX3-FOXO1 is now known to drive cancer-promoting gene expression programs through creation of distant genetic elements called super enhancers.
An ossifying fibromyxoid tumor is a type of myxoma. It presents in the extremities more frequently than the trunk. It is derived from mesenchyme. Appearance in the head and neck is rare, but has been reported. Its malignancy has been characterized as "intermediate".

Gliosarcoma is a rare type of glioma, a cancer of the brain that comes from glial, or supportive, brain cells, as opposed to the neural brain cells. Gliosarcoma is a malignant cancer, and is defined as a glioblastoma consisting of gliomatous and sarcomatous components. Primary gliosarcoma (PGS) is classified as a grade IV tumor and a subtype of glioblastoma multiforme in the 2007 World Health Organization classification system (GBM). Because of a lack of specific and clear diagnostic criteria, the word "gliosarcoma" was frequently used to refer to glial tumours with mesenchymal properties, such as the ability to make collagen and reticulin.
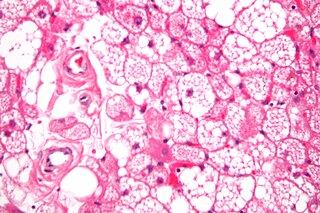
A hibernoma is a benign neoplasm of vestigial brown fat. The term was originally used by the French anatomist Louis Gery in 1914.

A Neurothekeoma (NT) is a type of rare benign cutaneous tumor that usually develops on the head and neck. They often occur in the second and early third decades of life and tend to afflict women more frequently than men. First described by Gallager and Helwig, who proposed the term in order to reflect the presumed origin of the lesion from nerve sheath. Microscopically, the lesions described closely resembled the tumor, "nerve sheath myxoma (NSM)", an entity first described by Harkin and Reed. The latter had, through the years, been variously described as "bizarre cutaneous neurofibroma", "myxoma of nerve sheath", and "pacinian neurofibroma".

Fetal adenocarcinoma (FA) of the lung is a rare subtype of pulmonary adenocarcinoma that exhibits tissue architecture and cell characteristics that resemble fetal lung tissue upon microscopic examination. It is currently considered a variant of solid adenocarcinoma with mucin production.

Giant-cell carcinoma of the lung (GCCL) is a rare histological form of large-cell lung carcinoma, a subtype of undifferentiated lung cancer, traditionally classified within the non-small-cell lung carcinomas (NSCLC).

Myoepithelioma of the head and neck, also myoepithelioma, is a salivary gland tumour of the head and neck that is usually benign. When malignant, which is exceedingly rare, they are known as malignant myoepithelioma or Myoepithelial carcinoma, and they account for 1% of the salivary tumors with poor prognosis.
A rhabdomyoblast is a cell type which is found in some rhabdomyosarcomas. When found histologically, a rhabdomyoblast aids the diagnosis of embryonal, alveolar, spindle cell/sclerosing, and pleomorphic rhabdomyosarcomas; however, in a tumor, expression of the rhabdomyoblast phenotype is not the only factor in diagnosing a rhabdomyosarcoma. Mesenchymal malignancies can exhibit this phenotype as well. Immunohistochemistry techniques allow for the sensitive detection of desmin, vimentin, muscle specific actin, and MyoD1. Similarly the rhabdomyoblast phenotype can be detected morphologically. Rhabdomyoblasts are early stage mesenchymal cells, having the potential to differentiate into a wide range of skeletal cells. Each stage of differentiation exhibits unique and distinguishable histological characteristics. In its initial from, stellate cells with amphiphilic cytoplasm and ovular central nuclei are observed. Commonly referred to as rhabdoid features, the maturing rhabdomyoblast will likely exhibit low levels of eosinophilic cytoplasm in proximal distances to the nucleus. As maturation and differentiation progress, the cell's cytoplasmic levels of white blood cells increase; additionally, elongated shapes, commonly depicted as “tadpole”, “strap” and "spider cells", are observed. In the concluding phase of differentiation, the white blood cell rich cytoplasm appears bright and exhibits cross-striation. The highly regulated organization of actin and myosin microfilaments in contractile proteins results in this appearance.
A sialoblastoma is a low-grade salivary gland neoplasm that recapitulates primitive salivary gland anlage. It has previously been referred to as congenital basal cell adenoma, embryoma, or basaloid adenocarcinoma. It is an extremely rare tumor, with less than 100 cases reported worldwide.
A ceruminous adenoma is a benign glandular neoplasm which arises from the ceruminous glands located within the external auditory canal. These glands are found within the outer one third to one half of the external auditory canal, more common along the posterior surface; therefore, the tumor develops within a very specific location.
Metanephric dysplastic hematoma of the sacral region (MDHSR) has been described by Cozzutto and Lazzaroni-Fossati in 1980, by Posalaki et al. in 1981 and by Cozzutto et al. in 1982. Three additional cases were seen by Finegold.

Sclerosing polycystic adenosis is a rare salivary gland tumor first described in 1996 by Dr. Brion Smith. The major salivary glands, specifically the parotid gland and the submandibular gland, are affected most commonly. Patients usually come to clinical attention with a mass or swelling in their salivary glands in the 5th decade of life, with females affected much more commonly than males. Nearly all of the cases reported so far have a benign behavior, although there is a single case that has had an associated malignant transformation.
Ectomesenchymal chondromyxoid tumor (ECT) is a benign intraoral tumor with presumed origin from undifferentiated (ecto)mesenchymal cells. There are some who think it is a myoepithelial tumor type.
Renal anaplastic sarcoma is a rare tumour of the kidney. By 2017 about 25 cases have been reported. This tumour occurs in children and young adults and is more common in females than males.
