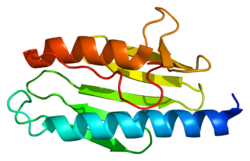
Frataxin is a protein that in humans is encoded by the FXNgene.[5][6]
It is located in the mitochondrion and Frataxin mRNA is mostly expressed in tissues with a high metabolic rate. The function of frataxin is not clear but it is involved in assembly of iron-sulfur clusters. It has been proposed to act as either an iron chaperone or an iron storage protein. Reduced expression of frataxin is the cause of Friedreich's ataxia.
Structure
X-ray crystallography has shown that human frataxin consists of a β-sheet that supports a pair of parallel α-helices, forming a compact αβ sandwich.[7] Frataxin homologues in other species are similar, sharing the same core structure. However, the frataxin tail sequences, extending from the end of one helix, diverge in sequence and differ in length. Human frataxin has a longer tail sequence than frataxin found in bacteria or yeast. It is hypothesized that the purpose of the tail is to stabilize the protein.[7]
Like most mitochondrial proteins, frataxin is synthesized in cytoplasmic ribosomes as large precursor molecules with mitochondrial targeting sequences. Upon entry into mitochondria, the molecules are broken down by a proteolytic reaction to yield mature frataxin.[8]
Function
Frataxin is localized to the mitochondrion. The function of frataxin is not entirely clear, but it seems to be involved in assembly of iron-sulfur clusters. It has been proposed to act as either an iron chaperone or an iron storage protein.[9]
Frataxin mRNA is predominantly expressed in tissues with a high metabolicrate (including liver, kidney, brown fat and heart). Mouse and yeast frataxin homologues contain a potential N-terminal mitochondrial targeting sequence, and human frataxin has been observed to co-localise with a mitochondrial protein. Furthermore, disruption of the yeast gene has been shown to result in mitochondrial dysfunction. Friedreich's ataxia is thus believed to be a mitochondrial disease caused by a mutation in the nuclear genome (specifically, expansion of an intronic GAA triplet repeat in the FXN gene, which encodes the protein frataxin.).[5][10][11]
Clinical significance
Reduced expression of frataxin is the cause of Friedreich's ataxia (FRDA), a neurodegenerative disease. The reduction in frataxin gene expression may be attributable from either the silencing of transcription of the frataxin gene because of epigenetic modifications in the chromosomal entity[12] or from the inability of splicing the expanded GAA repeats in the first intron of the pre-mRNA as seen in bacteria[13] and Human cells[14] or both. The expansion of intronic trinucleotide repeat GAA results in Friedreich's ataxia.[15] This expanded repeat causes R-loop formation, and using a repeat-targeted oligonucleotide to disrupt the R-loop can reactivate frataxin expression.[16]
96% of FRDA patients have a GAA trinucleotide repeat expansion in intron 1 of both alleles of their FXN gene.[17] Overall, this leads to a decrease in frataxin mRNA synthesis and a decrease (but not absence) in frataxin protein in people with FRDA. (A subset of FRDA patients have GAA expansion in one chromosome and a point mutation in the FXN exon in the other chromosome.) In the typical case, the length of the allele with the shorter GAA expansion inversely correlates with frataxin levels. FRDA patients' peripheral tissues typically have less than 10% of the frataxin levels exhibited by unaffected people.[17] Lower levels of frataxin result in earlier disease onset and faster progression.
FRDA is characterized by ataxia, sensory loss, and cardiomyopathy. The reason frataxin deficiency causes these symptoms is not entirely clear. On a cellular level, it is linked to iron accumulation in the mitochondria and increased oxidant sensitivity. For reasons that are not well understood, this primarily affects the tissue of the dorsal root ganglia, cerebellum, and heart muscle.[8]
Animal studies
In mice, complete inactivation of the FXN homolog (Frda) is lethal in the early embryonic stage.[18] Although nearly all organisms express a frataxin homologue, the GAA repeat in intron 1 only exists in humans and other primates, so the mutation that causes FDRA can't occur naturally in other animals. Scientists have developed several options to model this disease in mice. One approach is to silence frataxin expression in just one specific tissue type of interest: the heart (mice modified this way are called MCK), all neurons (NSE), or just the spinal cord and cerebellum (PRP).[19] Another approach involves inserting a GAA expansion into the first intron of the mouse FXN gene, which should inhibit frataxin production, just like in humans. Mice that are homozygous for this modified gene are called KIKI (knock-in knock-in), and the compound heterozygotes formed by crossing KIKI mice with frataxin knockout mice are called KIKO (knock-in knock-out). However, even KIKO mice still express 25-36% of the normal frataxin level, and show very mild symptoms. The final approach involves creating transgenic mice with a GAA-expanded version of the human frataxin gene. These mice are called YG22R (one GAA sequence of 190 repeats) and YG8R (two GAA sequences of 90 and 190 repeats). These mice show symptoms similar to human patients.[19]
An overexpression of frataxin in Drosophila has shown an increase in antioxidant capability, resistance to oxidative stress insults and longevity,[20] supporting the theory that the role of frataxin is to protect the mitochondria from oxidative stress and the ensuing cellular damage.
Fibroblasts from a mouse model of FRDA and FRDA patient fibroblasts show increased levels of DNA double-strand breaks.[21] A lentivirus gene delivery system was used to deliver the frataxin gene to the FRDA mouse model and human patient cells, and this resulted in long-term restored expression of frataxin mRNA and frataxin protein. This restored expression of the frataxin gene was accompanied by a substantial reduction in the number of DNA double-strand breaks.[21] The impaired frataxin in FRDA cells appears to cause reduced capacity for repair of DNA damage and this may contribute to neurodegeneration.[21]
Interactions
Frataxin has been shown to biologically interact with the enzyme PMPCB.[22]
↑Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). "Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin". Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID9241270. S2CID5883249.
↑Pan X, Ding Y, Shi L (Nov 2009). "The roles of SbcCD and RNaseE in the transcription of GAA x TTC repeats in Escherichia coli". DNA Repair. 8 (11): 1321–7. doi:10.1016/j.dnarep.2009.08.001. PMID19733517.
Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (Jun 1997). "Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin". Science. 276 (5319): 1709–12. doi:10.1126/science.276.5319.1709. PMID9180083.
Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). "Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin". Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID9241270. S2CID5883249.
Wilson RB, Roof DM (Aug 1997). "Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue". Nature Genetics. 16 (4): 352–7. doi:10.1038/ng0897-352. PMID9241271. S2CID22652291.
Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (Oct 1997). "Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia". Nature Genetics. 17 (2): 215–7. doi:10.1038/ng1097-215. PMID9326946. S2CID23151137.
Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M (1997). "Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo". Neurobiology of Disease. 4 (2): 103–13. doi:10.1006/nbdi.1997.0139. PMID9331900. S2CID6520439.
Zühlke C, Laccone F, Cossée M, Kohlschütter A, Koenig M, Schwinger E (Jul 1998). "Mutation of the start codon in the FRDA1 gene: linkage analysis of three pedigrees with the ATG to ATT transversion points to a unique common ancestor". Human Genetics. 103 (1): 102–5. doi:10.1007/s004390050791. PMID9737785. S2CID26999143.
Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, Kostrzewa M, Nivelon-Chevallier A, Gustavson KH, Kohlschütter A, Müller U, Mandel JL, Brice A, Koenig M, Cavalcanti F, Tammaro A, De Michele G, Filla A, Cocozza S, Labuda M, Montermini L, Poirier J, Pandolfo M (Feb 1999). "Friedreich's ataxia: point mutations and clinical presentation of compound heterozygotes". Annals of Neurology. 45 (2): 200–6. doi:10.1002/1531-8249(199902)45:2<200::AID-ANA10>3.0.CO;2-U. PMID9989622. S2CID24885238.
Coppola G, De Michele G, Cavalcanti F, Pianese L, Perretti A, Santoro L, Vita G, Toscano A, Amboni M, Grimaldi G, Salvatore E, Caruso G, Filla A (May 1999). "Why do some Friedreich's ataxia patients retain tendon reflexes? A clinical, neurophysiological and molecular study". Journal of Neurology. 246 (5): 353–7. doi:10.1007/s004150050362. PMID10399865. S2CID7367457.
Forrest SM, Knight M, Delatycki MB, Paris D, Williamson R, King J, Yeung L, Nassif N, Nicholson GA (Aug 1998). "The correlation of clinical phenotype in Friedreich ataxia with the site of point mutations in the FRDA gene". Neurogenetics. 1 (4): 253–7. doi:10.1007/s100480050037. PMID10732799. S2CID7463903.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.