In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
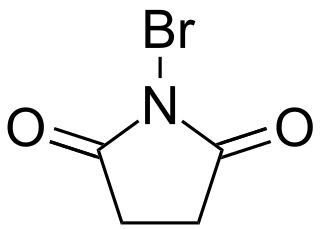
N-Bromosuccinimide or NBS is a chemical reagent used in radical substitution, electrophilic addition, and electrophilic substitution reactions in organic chemistry. NBS can be a convenient source of Br•, the bromine radical.
The Pictet–Spengler reaction is a chemical reaction in which a β-arylethylamine undergoes condensation with an aldehyde or ketone followed by ring closure. The reaction was first discovered in 1911 by Amé Pictet and Theodor Spengler. Traditionally an acidic catalyst in protic solvent was employed with heating, however the reaction has been shown to work in aprotic media in superior yields and sometimes without acid catalysis. The Pictet–Spengler reaction can be considered a special case of the Mannich reaction, which follows a similar reaction pathway. The driving force for this reaction is the electrophilicity of the iminium ion generated from the condensation of the aldehyde and amine under acid conditions. This explains the need for an acid catalyst in most cases, as the imine is not electrophilic enough for ring closure but the iminium ion is capable of undergoing the reaction.
The Bouveault aldehyde synthesis is a one-pot substitution reaction that replaces an alkyl or aryl halide with a formyl group using a N,N-disubstituted formamide. For primary alkyl halides this produces the homologous aldehyde one carbon longer. For aryl halides this produces the corresponding carbaldehyde. The Bouveault aldehyde synthesis is an example of a formylation reaction, and is named for French scientist Louis Bouveault.
The Reissert indole synthesis is a series of chemical reactions designed to synthesize indole or substituted-indoles from ortho-nitrotoluene 1 and diethyl oxalate 2.

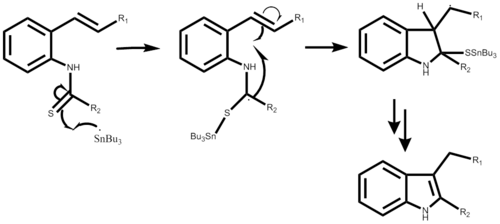
The Gassman indole synthesis is a series of chemical reactions used to synthesize substituted indoles by addition of an aniline and a ketone bearing a thioether substituent.
The Barton–McCombie deoxygenation is an organic reaction in which a hydroxy functional group in an organic compound is replaced by a hydrogen to give an alkyl group. It is named after British chemists Sir Derek Harold Richard Barton and Stuart W. McCombie.
Ring-closing metathesis (RCM) is a widely used variation of olefin metathesis in organic chemistry for the synthesis of various unsaturated rings via the intramolecular metathesis of two terminal alkenes, which forms the cycloalkene as the E- or Z- isomers and volatile ethylene.
The Dowd–Beckwith ring-expansion reaction is an organic reaction in which a cyclic β-keto ester is expanded by up to 4 carbons in a free radical ring expansion reaction through an α-alkylhalo substituent. The radical initiator system is based on AIBN and tributyltin hydride. The cyclic β-keto ester can be obtained through a Dieckmann condensation. The original reaction consisted of a nucleophilic aliphatic substitution of the enolate of ethyl cyclohexanone-2-carboxylate with 1,4-diiodobutane and sodium hydride followed by ring expansion to ethyl cyclodecanone-6-carboxylate. A side-reaction is organic reduction of the iodoalkane.

The Baker–Venkataraman rearrangement is the chemical reaction of 2-acetoxyacetophenones with base to form 1,3-diketones.

The Bartoli indole synthesis is the chemical reaction of ortho-substituted nitroarenes and nitrosoarenes with vinyl Grignard reagents to form substituted indoles.
The Castro–Stephens coupling is a cross coupling reaction between a copper(I) acetylide and an aryl halide in pyridine, forming a disubstituted alkyne and a copper(I) halide.

Tohru Fukuyama is a Japanese organic chemist and Professor of Chemistry at University of Tokyo in Japan. He discovered the Fukuyama coupling in 1998.

Indole is an aromatic, heterocyclic, organic compound with the formula C8H7N. It has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered pyrrole ring. Indole is widely distributed in the natural environment and can be produced by a variety of bacteria. As an intercellular signal molecule, indole regulates various aspects of bacterial physiology, including spore formation, plasmid stability, resistance to drugs, biofilm formation, and virulence. The amino acid tryptophan is an indole derivative and the precursor of the neurotransmitter serotonin.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.

The Birch reduction is an organic reaction that is used to convert arenes to 1,4-Cyclohexadiene. The reaction is named after the Australian chemist Arthur Birch and involves the organic reduction of aromatic rings in an amine solvent with an alkali metal and a proton source. Unlike catalytic hydrogenation, Birch reduction does not reduce the aromatic ring all the way to a cyclohexane.
Radical cyclization reactions are organic chemical transformations that yield cyclic products through radical intermediates. They usually proceed in three basic steps: selective radical generation, radical cyclization, and conversion of the cyclized radical to product.
The imine Diels–Alder reaction involves the transformation of all-carbon dienes and imine dienophiles into tetrahydropyridines.
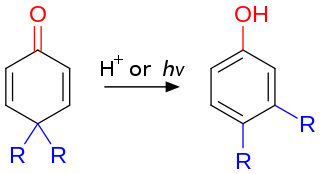
The dienone–phenol rearrangement is a reaction in organic chemistry first reported in 1921 by Karl von Auwers and Karl Ziegler. A common example of dienone–phenol rearrangement is 4,4-disubstituted cyclohexadienone converting into a stable 3,4-disubstituted phenol in presence of acid. A similar rearrangement is possible with a 2,2-disubstituted cyclohexadienone to its corresponding disubstituted phenol. Usually this type of rearrangement is spontaneous unless a dichloromethyl group is present at the 4th position or the process is otherwise blocked.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :