A glycosyl donor is a carbohydrate mono- or oligosaccharide that will react with a suitable glycosyl acceptor to form a new glycosidic bond. By convention, the donor is the member of this pair that contains the resulting anomeric carbon of the new glycosidic bond. [1] The resulting reaction is referred to as a glycosylation or chemical glycosylation.

A carbohydrate is a biomolecule consisting of carbon (C), hydrogen (H) and oxygen (O) atoms, usually with a hydrogen–oxygen atom ratio of 2:1 (as in water) and thus with the empirical formula Cm(H2O)n (where m may be different from n). This formula holds true for monosaccharides. Some exceptions exist; for example, deoxyribose, a sugar component of DNA, has the empirical formula C5H10O4. The carbohydrates are technically hydrates of carbon; structurally it is more accurate to view them as aldoses and ketoses.
An oligosaccharide is a saccharide polymer containing a small number of monosaccharides. Oligosaccharides can have many functions including cell recognition and cell binding. For example, glycolipids have an important role in the immune response.
A glycosyl acceptor: is any suitable nucleophile-containing molecule that will react with a glycosyl donor to form a new glycosidic bond. By convention, the acceptor is the member of this pair which did not contain the resulting anomeric carbon of the new glycosidic bond. Since the nucleophilic atom of the acceptor is typically an oxygen atom, this can be remembered using the mnemonic of the acceptor is the alcohol. A glycosyl acceptor can be a mono- or oligosaccharide that contains an available nucleophile, such as an unprotected hydroxyl.
In a glycosyl donor, a leaving group is required at the anomeric position. The simplest leaving group is the OH group that is naturally present in monosaccharides, but it requires activation by acid catalysis in order to function as leaving group (in the Fischer glycosylation). More effective leaving groups are in general used in the glycosyl donors employed in chemical synthesis of glycosides. Typical leaving groups are halides, thioalkyl groups, or imidates, but acetate, phosphate, and O-pentenyl groups are also employed. Natural glycosyl donors contain phosphates as leaving groups. [1]
In chemistry, a leaving group is a molecular fragment that departs with a pair of electrons in heterolytic bond cleavage. Leaving groups can be anions or neutral molecules, but in either case it is crucial that the leaving group be able to stabilize the additional electron density that results from bond heterolysis. Common anionic leaving groups are halides such as Cl−, Br−, and I−, and sulfonate esters such as tosylate (TsO−). Fluoride (F−) functions as a leaving group in the nerve-agent sarin gas. Common neutral molecule leaving groups are water and ammonia. Leaving groups may also be positively charged cations (such as H+ released during the nitration of benzene); these are also known specifically as electrofuges.
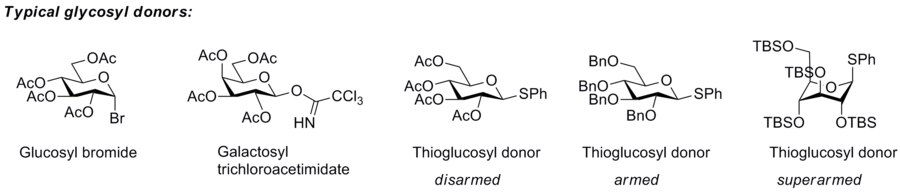
The so-called “armed-disarmed” principle
The concept of armed and disarmed glycosyl donors refers to the increased reactivity of benzylated over benzoylated glycosyl donors, a phenomenon observed very early, [2] and which originates from the greater electron-withdrawing capability of ester blocking groups over ether blocking groups. However, it was Bertram Fraser-Reid who realised that benzylated glycosyl donors can be activated when benzoylated donors are not, and invented the terms armed glycosyl donor for the former, and disarmed glycosyl donor for the latter. He and his group showed that armed glycosyl donors could be coupled to a glycosyl acceptor, that was at the same time a disarmed glycosyl donor, without self-coupling of the disarmed donor/acceptor. [3] This approach allowed him to carry out a one-pot synthesis of a trisaccharide by the n-pentenyl glycoside method. [4]

The armed/disarmed approach to glycosylation is an effective way to prevent sugar molecules from self-glycosylation when synthesizing disaccharides. This approach was first recognized when acetylated sugars only acted as glycosyl acceptors when reacted with benzylated sugars. The acetylated sugars were termed “disarmed” while the benzylated sugars were termed “armed”.
Bertram Oliver "Bert" Fraser-Reid is a Jamaican synthetic organic chemist who has been widely recognised for his work using carbohydrates as starting materials for chiral materials and on the role of oligosaccharides in immune response.
The concept has been extended to superarmed glycosyl donor by Mikael Bols and his collaborators. He realised that the hydroxy groups of carbohydrates are less electron-withdrawing towards the anomeric center when they are axial than when they are equatorial, which means that glycosyl donor conformers with more axial oxy functions are more reactive. [5] Protection of a glycosyl donor with bulky silyl groups (tert-butyldimethylsilyl or triisopropyl) cause it to change conformation to a more axial-rich conformation that, as a consequence, is more reactive, which Bols and his group called superarmed. They showed that a superarmed donor can be coupled to an armed glycosyl donor/acceptor. [6]
Mikael Bols is a synthetic organic chemist who is mainly known for his work on carbohydrates and artificial enzymes.
Silylation is the introduction of a (usually) substituted silyl group (R3Si) to a molecule. The process is the basis of organosilicon chemistry.