A glycosidic bond or glycosidic linkage is a type of ether bond that joins a carbohydrate (sugar) molecule to another group, which may or may not be another carbohydrate.

In chemistry, a glycoside is a molecule in which a sugar is bound to another functional group via a glycosidic bond. Glycosides play numerous important roles in living organisms. Many plants store chemicals in the form of inactive glycosides. These can be activated by enzyme hydrolysis, which causes the sugar part to be broken off, making the chemical available for use. Many such plant glycosides are used as medications. Several species of Heliconius butterfly are capable of incorporating these plant compounds as a form of chemical defense against predators. In animals and humans, poisons are often bound to sugar molecules as part of their elimination from the body.
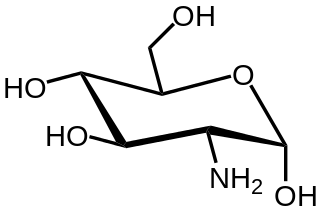
In organic chemistry, an amino sugar is a sugar molecule in which a hydroxyl group has been replaced with an amine group. More than 60 amino sugars are known, with one of the most abundant being N-Acetyl-d-glucosamine, which is the main component of chitin.
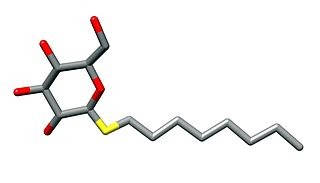
n-Octyl β-d-thioglucopyranoside is a mild nonionic detergent that is used for cell lysis or to solubilise membrane proteins without denaturing them. This is particularly of use in order to crystallise them or to reconstitute them into lipid bilayers. It has a critical micelle concentration of 9 mM.
Glycal is a name for cyclic enol ether derivatives of sugars having a double bond between carbon atoms 1 and 2 of the ring. The term "glycal" should not be used for an unsaturated sugar that has a double bond in any position other than between carbon atoms 1 and 2.
An Endoglycosidase is an enzyme that releases oligosaccharides from glycoproteins or glycolipids. It may also cleave polysaccharide chains between residues that are not the terminal residue, although releasing oligosaccharides from conjugated protein and lipid molecules is more common.
The Reformatsky reaction is an organic reaction which condenses aldehydes or ketones with α-halo esters using metallic zinc to form β-hydroxy-esters:

Glycosyltransferases are enzymes that establish natural glycosidic linkages. They catalyze the transfer of saccharide moieties from an activated nucleotide sugar to a nucleophilic glycosyl acceptor molecule, the nucleophile of which can be oxygen- carbon-, nitrogen-, or sulfur-based.
The Koenigs–Knorr reaction in organic chemistry is the substitution reaction of a glycosyl halide with an alcohol to give a glycoside. It is one of the oldest glycosylation reactions. It is named after Wilhelm Koenigs (1851–1906), a student of von Baeyer and fellow student with Hermann Emil Fischer, and Edward Knorr, a student of Koenigs.
The term glycosynthase refers to a class of proteins that have been engineered to catalyze the formation of a glycosidic bond. Glycosynthase are derived from glycosidase enzymes, which catalyze the hydrolysis of glycosidic bonds. They were traditionally formed from retaining glycosidase by mutating the active site nucleophilic amino acid to a small non-nucleophilic amino acid. More modern approaches use directed evolution to screen for amino acid substitutions that enhance glycosynthase activity.

Glycoside hydrolases catalyze the hydrolysis of glycosidic bonds in complex sugars. They are extremely common enzymes with roles in nature including degradation of biomass such as cellulose (cellulase), hemicellulose, and starch (amylase), in anti-bacterial defense strategies, in pathogenesis mechanisms and in normal cellular function. Together with glycosyltransferases, glycosidases form the major catalytic machinery for the synthesis and breakage of glycosidic bonds.
Nucleotide sugars are the activated forms of monosaccharides. Nucleotide sugars act as glycosyl donors in glycosylation reactions. Those reactions are catalyzed by a group of enzymes called glycosyltransferases.
Intramolecular aglycon delivery is a synthetic strategy for the construction of glycans. This approach is generally used for the formation of difficult glycosidic linkages.

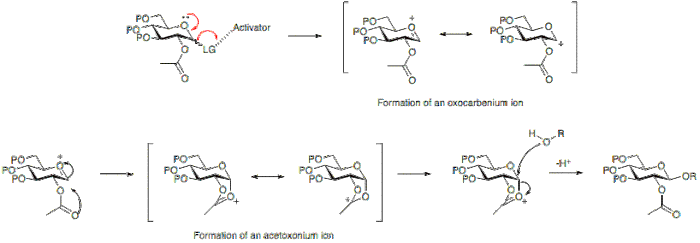
The armed/disarmed approach to glycosylation is an effective way to prevent sugar molecules from self-glycosylation when synthesizing disaccharides. This approach was first recognized when acetylated sugars only acted as glycosyl acceptors when reacted with benzylated sugars. The acetylated sugars were termed “disarmed” while the benzylated sugars were termed “armed”.
The Crich β-mannosylation in organic chemistry is a synthetic strategy which is used in carbohydrate synthesis to generate a 1,2-cis-glycosidic bond. This type of linkate is generally very difficult to make, and specific methods like the Crich β-mannosylation are used to overcome these issues.
A glycosyl donor is a carbohydrate mono- or oligosaccharide that will react with a suitable glycosyl acceptor to form a new glycosidic bond. By convention, the donor is the member of this pair that contains the resulting anomeric carbon of the new glycosidic bond. The resulting reaction is referred to as a glycosylation or chemical glycosylation.
A glycosyl acceptor is any suitable nucleophile-containing molecule that will react with a glycosyl donor to form a new glycosidic bond. By convention, the acceptor is the member of this pair which did not contain the resulting anomeric carbon of the new glycosidic bond. Since the nucleophilic atom of the acceptor is typically an oxygen atom, this can be remembered using the mnemonic of the acceptor is the alcohol. A glycosyl acceptor can be a mono- or oligosaccharide that contains an available nucleophile, such as an unprotected hydroxyl.
Carbohydrate synthesis is a sub-field of organic chemistry concerned specifically with the generation of natural and unnatural carbohydrate structures. This can include the synthesis of monosaccharide residues or structures containing more than one monosaccharide, known as oligosaccharides.

Glucanases are enzymes that break down large polysaccharides via hydrolysis. The product of the hydrolysis reaction is called a glucan, a linear polysaccharide made of up to 1200 glucose monomers, held together with glycosidic bonds. Glucans are abundant in the endosperm cell walls of cereals such as barley, rye, sorghum, rice, and wheat. Glucanases are also referred to as lichenases, hydrolases, glycosidases, glycosyl hydrolases, and/or laminarinases. Many types of glucanases share similar amino acid sequences but vastly different substrates. Of the known endo-glucanases, 1,3-1,4-β-glucanase is considered the most active.

Trichloroacetonitrile is an organic compound with the formula CCl3CN. It is a colourless liquid, although commercial samples often are brownish. It is used commercially as a precursor to the fungicide etridiazole. It is prepared by dehydration of trichloroacetamide. As a bifunctional compound, trichloroacetonitrile can react at both the trichloromethyl and the nitrile group. The electron-withdrawing effect of the trichloromethyl group activates the nitrile group for nucleophilic additions. The high reactivity makes trichloroacetonitrile a versatile reagent, but also causes its susceptibility towards hydrolysis.