Related Research Articles

In chemistry, a hydride is formally the anion of hydrogen (H−), a hydrogen atom with two electrons. The term is applied loosely. At one extreme, all compounds containing covalently bound H atoms are called hydrides: water (H2O) is a hydride of oxygen, ammonia is a hydride of nitrogen, etc. For inorganic chemists, hydrides refer to compounds and ions in which hydrogen is covalently attached to a less electronegative element. In such cases, the H centre has nucleophilic character, which contrasts with the protic character of acids. The hydride anion is very rarely observed.

Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
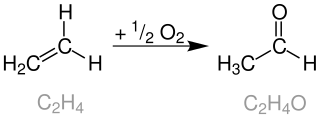
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.

Organotin chemistry is the scientific study of the synthesis and properties of organotin compounds or stannanes, which are organometallic compounds containing tin–carbon bonds. The first organotin compound was diethyltin diiodide, discovered by Edward Frankland in 1849. The area grew rapidly in the 1900s, especially after the discovery of the Grignard reagents, which are useful for producing Sn–C bonds. The area remains rich with many applications in industry and continuing activity in the research laboratory.
The Negishi coupling is a widely employed transition metal catalyzed cross-coupling reaction. The reaction couples organic halides or triflates with organozinc compounds, forming carbon-carbon bonds (C-C) in the process. A palladium (0) species is generally utilized as the metal catalyst, though nickel is sometimes used. A variety of nickel catalysts in either Ni0 or NiII oxidation state can be employed in Negishi cross couplings such as Ni(PPh3)4, Ni(acac)2, Ni(COD)2 etc.
Oppenauer oxidation, named after Rupert Viktor Oppenauer, is a gentle method for selectively oxidizing secondary alcohols to ketones.

Organocopper chemistry is the study of the physical properties, reactions, and synthesis of organocopper compounds, which are organometallic compounds containing a carbon to copper chemical bond. They are reagents in organic chemistry.
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formations.
Hydrosilylation, also called catalytic hydrosilation, describes the addition of Si-H bonds across unsaturated bonds. Ordinarily the reaction is conducted catalytically and usually the substrates are unsaturated organic compounds. Alkenes and alkynes give alkyl and vinyl silanes; aldehydes and ketones give silyl ethers. Hydrosilylation has been called the "most important application of platinum in homogeneous catalysis."
A carbometallation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond. The resulting carbon-metal bond can undergo further carbometallation reactions or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometallations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometallation.
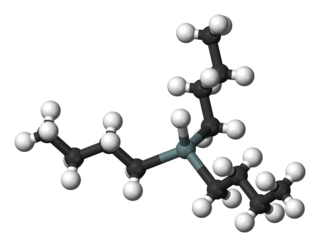
Tributyltin hydride is an organotin compound with the formula (C4H9)3SnH. It is a colorless liquid that is soluble in organic solvents. The compound is used as a source of hydrogen atoms in organic synthesis.
Metal carbon dioxide complexes are coordination complexes that contain carbon dioxide ligands. Aside from the fundamental interest in the coordination chemistry of simple molecules, studies in this field are motivated by the possibility that transition metals might catalyze useful transformations of CO2. This research is relevant both to organic synthesis and to the production of "solar fuels" that would avoid the use of petroleum-based fuels.
The Saegusa–Ito oxidation is a chemical reaction used in organic chemistry. It was discovered in 1978 by Takeo Saegusa and Yoshihiko Ito as a method to introduce α-β unsaturation in carbonyl compounds. The reaction as originally reported involved formation of a silyl enol ether followed by treatment with palladium(II) acetate and benzoquinone to yield the corresponding enone. The original publication noted its utility for regeneration of unsaturation following 1,4-addition with nucleophiles such as organocuprates.
Dehydrogenation of amine-boranes or dehydrocoupling of amine-boranes is a chemical process in main group and organometallic chemistry wherein dihydrogen is released by the coupling of two or more amine-borane adducts. This process is of due to the potential of using amine-boranes for hydrogen storage.
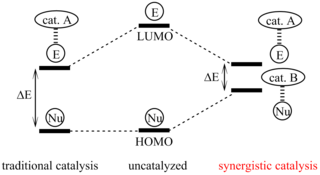
Synergistic catalysis is a specialized approach to catalysis whereby at least two different catalysts act on two different substrates simultaneously to allow reaction between the two activated materials. While a catalyst works to lower the energy of reaction overall, a reaction using synergistic catalysts work together to increase the energy level of HOMO of one of the molecules and lower the LUMO of another. While this concept has come to be important in developing synthetic pathways, this strategy is commonly found in biological systems as well.
Ionic hydrogenation refers to hydrogenation achieved by the addition of a hydride to substrate that has been activated by an electrophile. Some ionic hydrogenations entail addition of H2 to the substrate and some entail replacement of a heteroatom with hydride. Traditionally, the method was developed for acid-induced reductions with hydrosilanes. Alternatively ionic hydrogenation can be achieved using H2. Ionic hydrogenation is employed when the substrate can produce a stable carbonium ion. Polar double bonds are favored substrates.
In organic chemistry, the Fujiwara–Moritani reaction is a type of cross coupling reaction where an aromatic C-H bond is directly coupled to an olefinic C-H bond, generating a new C-C bond. This reaction is performed in the presence of a transition metal, typically palladium. The reaction was discovered by Yuzo Fujiwara and Ichiro Moritani in 1967. An external oxidant is required to this reaction to be run catalytically. Thus, this reaction can be classified as a C-H activation reaction, an oxidative Heck reaction, and a C-H olefination. Surprisingly, the Fujiwara–Moritani reaction was discovered before the Heck reaction.
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.
References
- ↑ Smith, Nicholas D.; Mancuso, John; Lautens, Mark (2000). "Metal-Catalyzed Hydrostannations". Chemical Reviews. 100 (8): 3257–3282. doi:10.1021/cr9902695. PMID 11749320.