Related Research Articles

A lysosome is a single membrane-bound organelle found in many animal cells. They are spherical vesicles that contain hydrolytic enzymes that digest many kinds of biomolecules. A lysosome has a specific composition, of both its membrane proteins and its lumenal proteins. The lumen's pH (~4.5–5.0) is optimal for the enzymes involved in hydrolysis, analogous to the activity of the stomach. Besides degradation of polymers, the lysosome is involved in cell processes of secretion, plasma membrane repair, apoptosis, cell signaling, and energy metabolism.

Lysosomal storage diseases are a group of over 70 rare inherited metabolic disorders that result from defects in lysosomal function. Lysosomes are sacs of enzymes within cells that digest large molecules and pass the fragments on to other parts of the cell for recycling. This process requires several critical enzymes. If one of these enzymes is defective due to a mutation, the large molecules accumulate within the cell, eventually killing it.
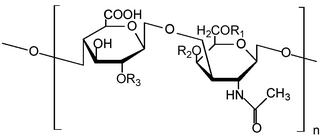
Glycosaminoglycans (GAGs) or mucopolysaccharides are long, linear polysaccharides consisting of repeating disaccharide units. The repeating two-sugar unit consists of a uronic sugar and an amino sugar, except in the case of the sulfated glycosaminoglycan keratan, where, in place of the uronic sugar there is a galactose unit. GAGs are found in vertebrates, invertebrates and bacteria. Because GAGs are highly polar molecules and attract water; the body uses them as lubricants or shock absorbers.
The terms glycans and polysaccharides are defined by IUPAC as synonyms meaning "compounds consisting of a large number of monosaccharides linked glycosidically". However, in practice the term glycan may also be used to refer to the carbohydrate portion of a glycoconjugate, such as a glycoprotein, glycolipid, or a proteoglycan, even if the carbohydrate is only an oligosaccharide. Glycans usually consist solely of O-glycosidic linkages of monosaccharides. For example, cellulose is a glycan composed of β-1,4-linked D-glucose, and chitin is a glycan composed of β-1,4-linked N-acetyl-D-glucosamine. Glycans can be homo- or heteropolymers of monosaccharide residues, and can be linear or branched.

Mucolipidosis is a group of inherited metabolic disorders that affect the body's ability to carry out the normal turnover of various materials within cells.

β-Glucocerebrosidase is an enzyme with glucosylceramidase activity that cleaves by hydrolysis the β-glycosidic linkage of the chemical glucocerebroside, an intermediate in glycolipid metabolism that is abundant in cell membranes. It is localized in the lysosome, where it remains associated with the lysosomal membrane. β-Glucocerebrosidase is 497 amino acids in length and has a molecular mass of 59,700 Da.

Pseudo-Hurler polydystrophy, also referred to as mucolipidosis III, is a lysosomal storage disease closely related to I-cell disease. This disorder is called Pseudo-Hurler because it resembles a mild form of Hurler syndrome, one of the mucopolysaccharide (MPS) diseases.

Insulin-like growth factor 2 receptor (IGF2R), also called the cation-independent mannose-6-phosphate receptor (CI-MPR) is a protein that in humans is encoded by the IGF2R gene. IGF2R is a multifunctional protein receptor that binds insulin-like growth factor 2 (IGF2) at the cell surface and mannose-6-phosphate (M6P)-tagged proteins in the trans-Golgi network.

Mannose-6-phosphate (M6P) is a molecule bound by lectin in the immune system. M6P is converted to fructose 6-phosphate by mannose phosphate isomerase.
The mannose 6-phosphate receptors (MPRs) are transmembrane glycoproteins that target enzymes to lysosomes in vertebrates.
N-acetylglucosamine-1-phosphate transferase (GlcNAc-1-phosphotransferase) is a transferase enzyme.

In enzymology, N-acetylglucosamine-6-phosphate deacetylase (EC 3.5.1.25), also known as GlcNAc-6-phosphate deacetylase or NagA, is an enzyme that catalyzes the deacetylation of N-acetylglucosamine-6-phosphate (GlcNAc-6-P) to glucosamine-6-phosphate (GlcN-6-P):
In enzymology, an UDP-N-acetylglucosamine—lysosomal-enzyme N-acetylglucosaminephosphotransferase is an enzyme that catalyzes the chemical reaction

UDP-N-acetylglucosamine—dolichyl-phosphate N-acetylglucosaminephosphotransferase is an enzyme that in humans is encoded by the DPAGT1 gene.

N-acetylglucosamine-1-phosphodiester alpha-N-acetylglucosaminidase is an enzyme that in humans is encoded by the NAGPA gene.
N-linked glycosylation is the attachment of an oligosaccharide, a carbohydrate consisting of several sugar molecules, sometimes also referred to as glycan, to a nitrogen atom, in a process called N-glycosylation, studied in biochemistry. The resulting protein is called an N-linked glycan, or simply an N-glycan.

In the fields of biochemistry and cell biology, the cation-dependent mannose-6-phosphate receptor (CD-MPR) also known as the 46 kDa mannose 6-phosphate receptor is a protein that in humans is encoded by the M6PR gene.
O-linked glycosylation is the attachment of a sugar molecule to the oxygen atom of serine (Ser) or threonine (Thr) residues in a protein. O-glycosylation is a post-translational modification that occurs after the protein has been synthesised. In eukaryotes, it occurs in the endoplasmic reticulum, Golgi apparatus and occasionally in the cytoplasm; in prokaryotes, it occurs in the cytoplasm. Several different sugars can be added to the serine or threonine, and they affect the protein in different ways by changing protein stability and regulating protein activity. O-glycans, which are the sugars added to the serine or threonine, have numerous functions throughout the body, including trafficking of cells in the immune system, allowing recognition of foreign material, controlling cell metabolism and providing cartilage and tendon flexibility. Because of the many functions they have, changes in O-glycosylation are important in many diseases including cancer, diabetes and Alzheimer's. O-glycosylation occurs in all domains of life, including eukaryotes, archaea and a number of pathogenic bacteria including Burkholderia cenocepacia, Neisseria gonorrhoeae and Acinetobacter baumannii.
GNPTG is a gene in the human body. It is one of three genes that were found to correlate with stuttering.
References
- ↑ "mucolipidosis II" at Dorland's Medical Dictionary
- ↑ Plante M, Claveau S, Lepage P, et al. (March 2008). "Mucolipidosis II: a single causal mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTAB) in a French Canadian founder population" (PDF). Clin. Genet. 73 (3): 236–44. doi:10.1111/j.1399-0004.2007.00954.x. PMID 18190596. S2CID 20999105.
- ↑ Bamshad, Lynn B. Jorde, John C. Carey, Michael J. (2010). Medical genetics (4th ed.). Philadelphia: Mosby/Elsevier. ISBN 9780323053730.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ Le, Tao (2014). First Aid for the USMLE 2014. New York: McGraw Hill Education. p. 77. ISBN 9780071831420.
- 1 2 Champe, Pamela (2004). Lippincott's Illustrated Reviews: Biochemistry. Richard A Harvey, Denise R Ferrier (3rd ed.). Philadelphia, Pa.: Lippincott-Raven. p. 167. ISBN 978-0-7817-2265-0.
- ↑ Tiede S, Storch S, Lübke T, et al. (2005). "Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase". Nat. Med. 11 (10): 1109–12. doi:10.1038/nm1305. PMID 16200072. S2CID 24959938.
- ↑ "Sahha.gov.mt - 2006 Dec;29_1". Archived from the original on 2012-02-26. Retrieved 2009-09-16.
- 1 2 3 4 5 "I Cell Disease - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 2017-11-02.
- ↑ "Inherited Metabolic Storage Diseases and BMT - MED - PEDS - Blood and Marrow Transplantation Program, University of Minnesota". Archived from the original on 2010-06-20. Retrieved 2009-12-01.
- ↑ "Yash Gandhi Foundation". Yash Gandhi Foundation. Retrieved 2023-09-25.