
Arthrogryposis (AMC) describes congenital joint contracture in two or more areas of the body. It derives its name from Greek, literally meaning 'curving of joints'.

Nijmegen breakage syndrome (NBS) is a rare autosomal recessive congenital disorder causing chromosomal instability, probably as a result of a defect in the double Holliday junction DNA repair mechanism and/or the synthesis dependent strand annealing mechanism for repairing double strand breaks in DNA.
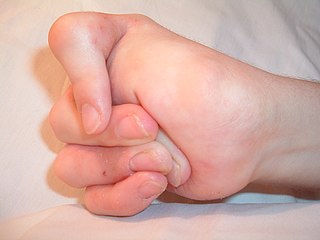
Freeman–Sheldon syndrome (FSS) is a very rare form of multiple congenital contracture (MCC) syndromes (arthrogryposes) and is the most severe form of distal arthrogryposis (DA). It was originally described by Ernest Arthur Freeman and Joseph Harold Sheldon in 1938.

Laurence–Moon syndrome (LMS) is a rare autosomal recessive genetic disorder associated with retinitis pigmentosa, spastic paraplegia, and mental disabilities.

CHIME syndrome, also known as Zunich–Kaye syndrome or Zunich neuroectodermal syndrome, is a rare congenital ichthyosis first described in 1983. The acronym CHIME is based on its main symptoms: colobomas, heart defects, ichthyosiform dermatosis, intellectual disability, and either ear defects or epilepsy. It is a congenital syndrome with only a few cases studied and published.
Renal dysplasia-limb defects syndrome, also known as Ulbright–Hodes syndrome, is a very rare autosomal recessive congenital disorder. It has been described in three infants, all of whom died shortly after birth.
CAMFAK syndrome is an acronym used to describe a rare inherited neurologic disease, characterized by peripheral and central demyelination of nerves, similar to that seen in Cockayne syndrome. The name "CAMFAK" comes from the first letters of the characteristic findings of the disease: cataracts, microcephaly, failure to thrive, and kyphoscoliosis. The disease may occur with or without failure to thrive and arthrogryposis.

Nucleoporin GLE1 is a protein that in humans is encoded by the GLE1 gene on chromosome 9.
Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Boomerang dysplasia is a lethal form of osteochondrodysplasia known for a characteristic congenital feature in which bones of the arms and legs are malformed into the shape of a boomerang. Death usually occurs in early infancy due to complications arising from overwhelming systemic bone malformations.
EEM syndrome is an autosomal recessive congenital malformation disorder affecting tissues associated with the ectoderm, and also the hands, feet and eyes.
Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy–Walker malformation and agenesis of corpus callosum.
Hydrolethalus syndrome (HLS) is a rare genetic disorder that causes improper fetal development, resulting in birth defects and, most commonly, stillbirth.

Hydrops-ectopic calcification-moth-eaten skeletal dysplasia is a defect in cholesterol biosynthesis. Greenberg characterized the condition in 1988.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
A Finnish heritage disease is a genetic disease or disorder that is significantly more common in people whose ancestors were ethnic Finns, natives of Finland and Northern Sweden (Meänmaa) and Northwest Russia. There are 36 rare diseases regarded as Finnish heritage diseases. The diseases are not restricted to Finns; they are genetic diseases with far wider distribution in the world, but due to founder effects and genetic isolation they are more common in Finns.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.
Lethal arthrogryposis with anterior horn cell disease (LAAHD) is an autosomal recessive genetic disorder characterized by reduced mobility of the foetus and early death.
Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.
Bruck syndrome is characterized as the combination of arthrogryposis multiplex congenita and osteogenesis imperfecta. Both diseases are uncommon, but concurrence is extremely rare which makes Bruck syndrome very difficult to research. Bruck syndrome is thought to be an atypical variant of osteogenesis imperfecta most resembling type III, if not its own disease. Multiple gene mutations associated with osteogenesis imperfecta are not seen in Bruck syndrome. Many affected individuals are within the same family, and pedigree data supports that the disease is acquired through autosomal recessive inheritance. Bruck syndrome has features of congenital contractures, bone fragility, recurring bone fractures, flexion joint and limb deformities, pterygia, short body height, and progressive kyphoscoliosis. Individuals encounter restricted mobility and pulmonary function. A reduction in bone mineral content and larger hydroxyapatite crystals are also detectable Joint contractures are primarily bilateral and symmetrical, and most prone to ankles. Bruck syndrome has no effect on intelligence, vision, or hearing.
