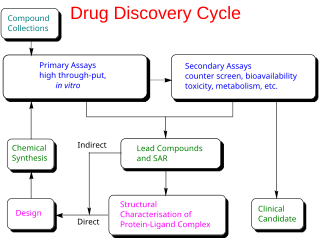
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
In the physical sciences, a partition coefficient (P) or distribution coefficient (D) is the ratio of concentrations of a compound in a mixture of two immiscible solvents at equilibrium. This ratio is therefore a comparison of the solubilities of the solute in these two liquids. The partition coefficient generally refers to the concentration ratio of un-ionized species of compound, whereas the distribution coefficient refers to the concentration ratio of all species of the compound.

In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from Latin ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.
Hit to lead (H2L) also known as lead generation is a stage in early drug discovery where small molecule hits from a high throughput screen (HTS) are evaluated and undergo limited optimization to identify promising lead compounds. These lead compounds undergo more extensive optimization in a subsequent step of drug discovery called lead optimization (LO). The drug discovery process generally follows the following path that includes a hit to lead stage:
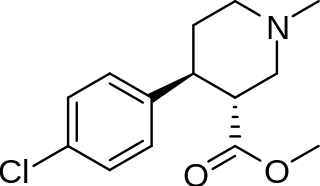
(+)-CPCA is a stimulant drug similar in structure to pethidine and to RTI-31, but nocaine is lacking the two-carbon bridge of RTI-31's tropane skeleton. This compound was first developed as a substitute agent for cocaine.
Druglikeness is a qualitative concept used in drug design for how "druglike" a substance is with respect to factors like bioavailability. It is estimated from the molecular structure before the substance is even synthesized and tested. A druglike molecule has properties such as:

Epiboxidine is a chemical compound which acts as a partial agonist at neural nicotinic acetylcholine receptors, binding to both the α3β4 and the α4β2 subtypes. It was developed as a less toxic analogue of the potent frog-derived alkaloid epibatidine, which is around 200 times stronger than morphine as an analgesic but produces extremely dangerous toxic nicotinic side effects.

Fluorenol, also known as hydrafinil, is an alcohol derivative of fluorene. In the most significant isomer, fluoren-9-ol or 9-hydroxyfluorene, the hydroxy group is located on the bridging carbon between the two benzene rings. Hydroxyfluorene can be converted to fluorenone by oxidation. It is a white-cream colored solid at room temperature.

AMDA (9-Aminomethyl-9,10-dihydroanthracene) is an organic compound which acts as a potent and selective antagonist for the 5-HT2A receptor. It has been used to help study the shape of the 5-HT2A protein, and develop a large family of related derivatives with even higher potency and selectivity.
CCR5 receptor antagonists are a class of small molecules that antagonize the CCR5 receptor. The C-C motif chemokine receptor CCR5 is involved in the process by which HIV, the virus that causes AIDS, enters cells. Hence antagonists of this receptor are entry inhibitors and have potential therapeutic applications in the treatment of HIV infections.
Relief from chronic pain remains a recognized unmet medical need. Consequently, the search for new analgesic agents is being intensively studied by the pharmaceutical industry. The TRPV1 receptor is a ligand gated ion channel that has been implicated in mediation of many types of pain and therefore studied most extensively. The first competitive antagonist, capsazepine, was first described in 1990; since then, several TRPV1 antagonists have entered clinical trials as analgesic agents. Should these new chemical entities relieve symptoms of chronic pain, then this class of compounds may offer one of the first novel mechanisms for the treatment of pain in many years.

8-Carboxamidocyclazocine (8-CAC) is an opioid analgesic drug related to cyclazocine, discovered by medicinal chemist Mark P. Wentland and co-workers in Cogswell Laboratory at Rensselaer Polytechnic Institute. Similarly to cyclazocine, 8-CAC acts as an agonist at both the μ and κ opioid receptors, but has a much longer duration of action than cyclazocine, and does not have μ antagonist activity. Unexpectedly it was discovered that the phenolic hydroxyl group of cyclazocine could be replaced by a carboxamido group with only slight loss of potency at opioid receptors, and this discovery has subsequently been used to develop many novel opioid derivatives where the phenolic hydroxy group has been replaced by either carboxamide or a variety of larger groups. Due to their strong κ-opioid agonist activity, these drugs are not suited for use as analgesics in humans, but have instead been researched as potential drugs for the treatment of cocaine addiction.
Ligand efficiency is a measurement of the binding energy per atom of a ligand to its binding partner, such as a receptor or enzyme.

VU-0238429 is a drug which acts as a selective positive allosteric modulator for the muscarinic acetylcholine receptor M5. It was the first selective ligand developed for the M5 subtype, and is structurally derived from older M1-selective positive allosteric modulators such as VU-0119498. Replacing the O-methyl- by a phenyl group further improves the receptor subtype selectivity.

Biphalin is a dimeric enkephalin endogenous peptide (Tyr-D-Ala-Gly-Phe-NH)2 composed of two tetrapeptides derived from enkephalins, connected 'tail-to-tail' by a hydrazide bridge. The presence of two distinct pharmacophores confers on biphalin a high affinity for both μ and δ opioid receptors (with an EC50 of about 1-5 nM for both μ and δ receptors), therefore it has analgesic activity. Biphalin presents a considerable antinociceptive profile. In fact, when administered intracerebroventricularly in mice, biphalin displays a potency almost 7-fold greater than that of the ultra-potent alkaloid agonist, etorphine and 7000-fold greater than morphine; biphalin and morphine were found to be equipotent after intraperitoneal administration. The extraordinary in vivo potency shown by this compound is coupled with low side-effects, in particular, to produce no dependency in chronic use. For these reasons, several efforts have been carried out in order to obtain more information about structure-activity relationship (SAR). Results clearly indicate that, at least for μ receptor binding, the presence of two pharmacophores is not necessary; Tyr1 is indispensable for analgesic activity, while replacing Phe at the position 4 and 4' with non-aromatic, but lipophilic amino acids does not greatly change the binding properties and in general 4,4' positions are found to be important to design biphalin analogues with increased potency and modified μ/δ selectivity. The hydrazide linker is not fundamental for activity or binding, and it can be conveniently substituted by different conformationally constrained cycloaliphatic diamine linkers.
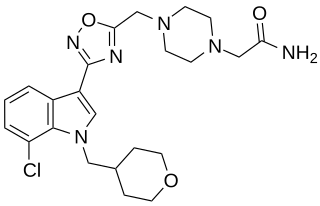
LBP-1 is a drug originally developed by Organon for the treatment of neuropathic pain, It acts as a potent and selective cannabinoid receptor agonist, with high potency at both the CB1 and CB2 receptors, but low penetration of the blood–brain barrier. This makes LBP-1 peripherally selective, and while it was effective in animal models of neuropathic pain and allodynia, it did not produce cannabinoid-appropriate responding suggestive of central effects, at any dose tested.

4-Fluoromethylphenidate is a stimulant drug that acts as a higher potency dopamine reuptake inhibitor than the closely related methylphenidate.

QMPSB is an arylsulfonamide-based synthetic cannabinoid that has been sold as a designer drug.
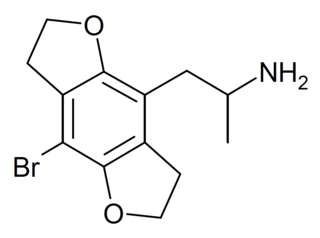
DOB-FLY is a recreational designer drug with psychedelic effects. It can be regarded as the alpha-methyl derivative of 2C-B-FLY or the partially saturated counterpart of bromo-dragonfly. Unlike bromo-dragonfly, DOB-FLY is only slightly more potent than DOB itself, with an active dose in humans of around 1 mg.


